
Mechanical stability of zinc oxide nanowires under tensile loading: is wurtzite stable at the nanoscale?†
Abstract
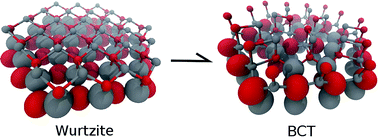
The mechanical response of zinc oxide nanowires under uniaxial tensile loading is investigated by molecular dynamics and supported by density functional calculations. Previous theoretical works predict a stress-induced phase transition which has not been observed experimentally in zinc oxide nanowires up to date. Here, we report an explanation for such a discrepancy. Our simulations reveal brittle failure at room temperature without phase transformation, in agreement with experiments. Interestingly, we also find that if the temperature is raised to 600 K, the phase transition occurs. A detailed reaction mechanism is proposed. For the first time, the associated rate constant has been calculated. Based on these results we propose an experimental procedure to finally observe the predicted phase transformation.
 Please wait while we load your content...
Please wait while we load your content...

