
Molecular design of N–NO2 substituted cycloalkanes derivatives Cm(N–NO2)m for energetic materials with high detonation performance and low impact sensitivity†
Yan-Yan Guoa,
Wei-Jie Chia,
Ze-Sheng Li*ab and
Quan-Song Li*a
aBeijing Key Laboratory of Photoelectronic/Electrophotonic Conversion Materials, Key Laboratory of Cluster Science of Ministry of Education, Beijing Key Laboratory for Chemical Power Source and Green Catalysis, School of Chemistry, Beijing Institute of Technology, Beijing 100081, China
bThe Academy of Fundamental and Interdisciplinary Sciences, Harbin Institute of Technology, Harbin 150080, China. E-mail: zeshengli@bit.edu.cn; liquansong@bit.edu.cn
First published on 20th April 2015
Abstract
For novel high-energy low-sensitivity energetic materials, a series of novel cycloalkanes derivatives Cm(N–NO2)m (m = 3–8) were theoretically designed by substitution of the hydrogen atoms with N–NO2 group. Density functional theory (DFT) calculations in combination with the isodesmic reaction and the Kamlet–Jacobs equations were employed to predict the heats of formation (HOFs) and the detonation properties. We found that the designed compounds have large positive HOFs, which are proportional to the amount of N–NO2 groups. Importantly, these compounds possess high crystal densities (1.85–1.95 g cm−3) and heats of detonation (1811–2054 kJ g−1), which lead to remarkable detonation properties (detonation velocities = 9.37–9.61 km s−1 and detonation pressures = 38.03–42.48 GPa) that are greater than those of the well-known energetic compounds 2,4,6,8,10,12-hexanitro-2,4,6,8,10,12-hexaazaisowurtzitane (CL-20), 1,3,5-trinitro-1,3,5-triazinane (RDX), and 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX). Moreover, the bond dissociation energy and the impact sensitivity index h50 values suggest that the title molecules are less sensitive than CL-20, and comparable to HMX and RDX. Therefore, our results show that the designed compounds may be promising candidates for energetic materials with notable detonation performance and low impact sensitivity.
1 Introduction
High energy density compounds (HEDCs) are special materials that store large quantities of energy and are used extensively for military purposes and civilian applications.1–5 In the past decades, great efforts have been made on designing and synthesizing all types of HEDCs, including all-nitrogen molecules, heterocyclic compounds, nitramines, prismane derivatives, cubane derivatives, nitro-triaziridine derivatives, and so on.6–10 For instance, cyclic nitramine compounds including 1,3,5-trinitro-1,3,5-triazinane (RDX) and 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX)11 constitute a class of energetic compounds owing to the low sensitivity, but their detonation properties do not measure up to the HEDC's requirements.2,11–15 So far, the effective way of improving the detonation performance is introducing high-energy groups, such as –NO2, –N3, –CN, and so on.16,17 As a consequence, Gong and coworkers introduced![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NO2 groups on HMX and RDX, and the resulted derivatives exhibit evidently enhanced detonation performance.6 Additionally, Zamani et al. reported that –NHNO2 substituted borazine-based compounds containing –N3 groups display superior detonation performance than conventional nitramine compounds HMX and RDX.18
N–NO2 groups on HMX and RDX, and the resulted derivatives exhibit evidently enhanced detonation performance.6 Additionally, Zamani et al. reported that –NHNO2 substituted borazine-based compounds containing –N3 groups display superior detonation performance than conventional nitramine compounds HMX and RDX.18
An ideal energetic compound requires substantial detonation performance, good thermal stability, low sensitivity to external stimuli, and environmental safety. However, the demands of high energy and insensitivity are quite often contradictory to each other, making the development of novel HEDCs a challenging problem.1 Therefore, much attention has been paid on designing HEDCs that exhibit good balance between the sensitivity and the performance. One popular strategy for the design of HEDCs is incorporating both fuel and oxidizer properties into one single molecule.19 On the basis of this strategy, Zhang and Shreeve reported the synthesis and full characterization of 3,3′-dinitroamino-4,4′-azoxyfurazan and its nitrogen-rich salts, where diverse N–O moieties create a balance between the detonation properties and the impact and friction sensitivities.20 Similarly, a set of polydinitroaminoprismanes are predicted to be potential HEDCs with balanced detonation performance and impact sensitivity and stability.21 Recently, a new design strategy for high-energy low-sensitivity HEDCs has been put forward by combining oxygen balance equal to zero, a combination of nitro and amino groups, and N-oxide in one molecule.22 In a similar way, two tetrazole derivatives with fused energetic groups (catenated nitrogen atoms) and insensitive fragments (tetrazole and FOX-7 segments) exhibit eminent detonation properties and acceptable insensitivity.23 Additionally, the formation of intramolecular or intermolecular hydrogen bonds and enhancement of conjugated effect are often employed to improve the molecular stability.24
As important chemical products and materials, cycloalkanes are similar to alkanes in general physical properties, especially the good structure stability and various means of preparation and activation.25–28 The strain energy in small-rings and the stronger London forces arising from the ring shape that allows for larger areas of contact imply the cycloalkanes might be good precursors for HEDCs, which motivated people to design novel energetic compounds using small-ring cycloalkanes as the core. Although much has been explored about HEDCs with small rings including triaziridines29 and epoxyethanes,30,31 however, to the best of our knowledge, no systematic studies on cycloalkanes-based HEDCs have been reported.
In present work six novel cycloalkanes derivatives Cm(N–NO2)m (m = 3–8) were theoretically designed by substitution of the hydrogen atoms with N–NO2 group, aiming at looking for new HEDCs with better detonation performance and lower sensitivity compared with those currently used. The structures of the designed compounds are presented in Fig. 1, where the molecules are nominated as C3–C8 based on the carbon numbers of the ring. Density functional theory (DFT) has been employed to predict the detonation performance and the thermal stability. Quantitative criteria considering both detonation performance (crystal density ρ ≈ 1.9 g cm−3, detonation velocity D ≈ 9.0 km s−1, P ≈ 40.0 GPa) and the stability requirement (bond dissociation energy of the initial step in thermolysis BDE > 84 kJ mol−1, the impact sensitivity h50 > 12 cm) are employed to filter the candidates and identify potential HEDCs.32 It is expected that our results could provide some useful information for laboratory synthesis of novel HEDCs.
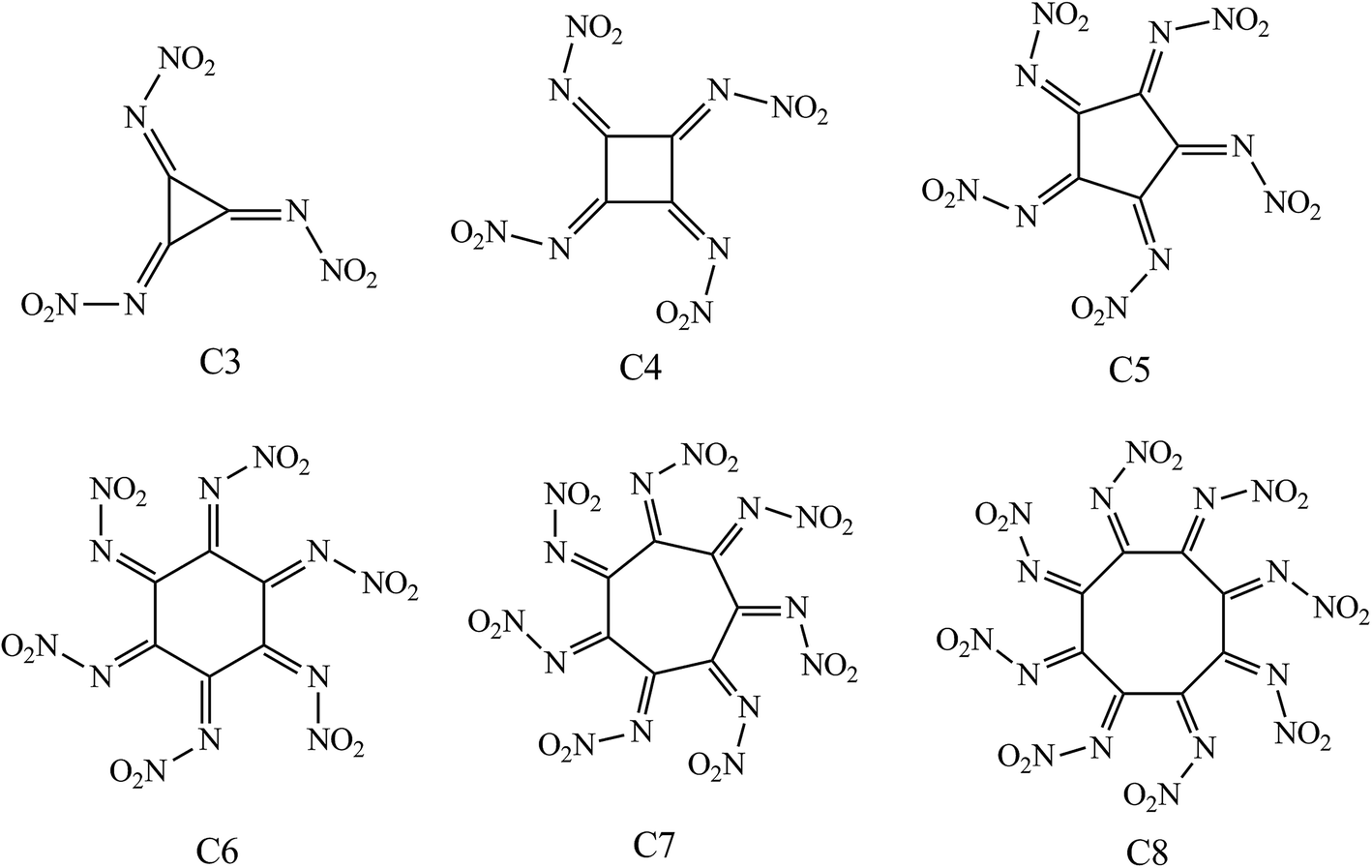 | ||
| Fig. 1 The molecular structure of the title compounds. | ||
2 Computational details
Previous studies have shown that the DFT-B3LYP33–35 method in combination with appropriate basis set is capable of figuring out the accurate energies, molecular structures, and vibrational frequencies of medium-sized molecules that are very close to the corresponding experimental results.36–38 In this paper, the geometry optimization of the molecular structure are carried out at B3LYP/6-31G**39,40 level with Gaussian 09 software package.41 The optimized structures are characterized to be energy minima on the potential energy surface by vibrational analysis without the presence of imaginary frequencies. The atoms coordinates of all designed compounds are listed (see Table S1–S6 in ESI†).The isodesmic reactions used to obtain the HOFgas of the designed compounds at 298 K are as follows:42,43
| Cm(N–NO2)m + 2mCH4 = CmH2m + m(CH3)2N–NO2, m = 3, 4, 5,6,7,8 | (1) |
N–NO2 groups in the designed compounds. For the isodesmic reaction, the heat of reaction (ΔrH298) at 298 K can be calculated from the following equation:| ΔrH298 = ∑ΔfHP − ∑ΔfHR = ΔE0 + ΔEZPE + ΔHT + ΔnRT | (2) |
For estimation of the detonation performance of the energetic compounds, it is also significant to calculate the HOF in solid state (HOFsolid) because it is directly related to the detonation characteristics. According to Hess' law,44 HOFsolid can be obtained by
| HOFsolid = HOFgas − HOFsub | (3) |
| HOFsub = αA2 + β(νσtotal2)1/2 + γ | (4) |
Detonation velocity (D) and detonation pressure (P), which are two important properties of energetic compounds, are estimated by the empirical Kamlet–Jacobs equations:49
D = 1.01(N![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) 0.5Q0.5)0.5(1 + 1.3ρ) 0.5Q0.5)0.5(1 + 1.3ρ)
| (5) |
|
P = 1.558ρ2N0.5Q0.5
| (6) |
is the average molecular weight of the gaseous products; Q is the heat of detonation (kJ g−1); ρ is the crystal density (g cm−3).
In addition, we obtained the crystal densities of the title compounds by using Materials Studio to predict the crystal structures.50 The Dreiding force field is capable of predicting the solid phase properties by searching the possible molecular packing among the most probable space groups.51
The heat of detonation Q and the volume V can be combined to obtain the value for the explosive power as shown in the following equation:52,53
| Explosive power = QV | (7) |
The value of the explosive power is compared with the explosive power of a standard explosive (picric acid, PAc) to obtain the power index, as shown in the following equation:
| Power index = (QV/QPAcVPAc) × 100 | (8) |
The thermal stabilities of the title compounds are evaluated by calculating the bond dissociation energy (BDE) of the N–NO2 bond. At 298 K, the BDE can be obtained via
| BDE0(A − B) = E0(A˙) + E0(B˙) − E0(A − B) | (9) |
The bond dissociation energy with ZPE correction can be calculated via
| BDE(A − B)ZPE = BDE0(A − B) + ΔEZPE | (10) |
The characteristic height (h50), which reflects the impact sensitivity and the stability of a compound,54,55 can be estimated using the following equation, as suggested by Cao:56
| h50 = 0.1296 + 98.64QNO22 − 0.03405OB100 | (11) |
OB100 can be calculated from the following equation:
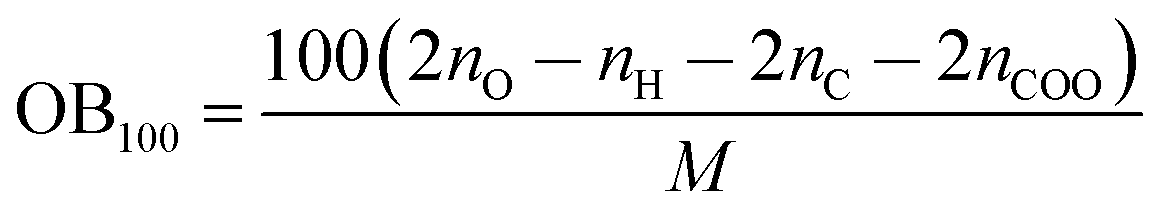 | (12) |
3 Results and discussion
3.1 Heat of formation
It is well-known that the heat of formation (HOF) is an important parameter to determine the detonation properties and energy content of an energetic compound. The total energies (E0), zero-point energies (EZPE), thermal corrections (HT), and the experimental HOFs data of the reference compounds57 are listed in Table 1. Table 2 summarizes E0, EZPE, HT, heats of formation in the gas phase (HOFgas), heats of sublimation (HOFsub) and heats of formation in solid phase (HOFsolid) of the title compounds at B3LYP/6-31G(d,p) level. As can be seen in Table 2, the HOFsolid of the title compounds are quite large positive values (676.63–1295.77 kJ mol−1), which meet the standard of energetic materials. The smallest HOFsolid is 676.73 kJ mol−1 of C3 among the designed compounds, which is much larger than that of RDX (70 kJ mol−1), HMX (75 kJ mol−1), and CL-20 (394.45 kJ mol−1).58 These high HOFsolid can be attributed to the presence of N–N and CN bonds and large strain energy of the ring. Besides, we can see the HOFsolid is strongly dependent on the number of CN–NO2 group. To clarify the dependence, the HOFsolid values as a function of the CN–NO2 amount are plotted in Fig. 2. Clearly, the HOFsolid values go up with the number increase of the CN–NO2 group and there exists a good linear relationship between them, which can be summarized as HOFsolid = 196.2 + 131.3n (R = 0.967). It is obvious that the change trend complies with the group additive rule, where the addition of each CN–NO2 group upturns the HOFsolid by 131.34 kJ mol−1.
| Compd | E0 (a.u.) | EZPE (a.u.) | HT (a.u.) | HOF (kJ mol−1) |
|---|---|---|---|---|
| CH4 | −40.524019 | 0.045026 | 0.003810 | −74.90 |
| N(CH3)2NO2 | −339.664624 | 0.095422 | 0.007624 | −5.02 |
| C3H6 | −117.904157 | 0.081475 | 0.004321 | 53.09 |
| C4H8 | −157.224450 | 0.111059 | 0.005071 | 28.42 |
| C5H10 | −196.571089 | 0.140993 | 0.006116 | −75.61 |
| C6H12 | −235.886784 | 0.170489 | 0.006950 | −122.31 |
| C7H14 | −275.203115 | 0.199394 | 0.007974 | −154.70 |
| C8H16 | −314.510007 | 0.228312 | 0.009142 | −124.50 |
| Compd | E0 (a.u.) | EZPE (a.u.) | HT (a.u.) | HOFgas (kJ mol−1) | HOFsub (kJ mol−1) | HOFsolid (kJ mol−1) |
|---|---|---|---|---|---|---|
| C3 | −893.630996 | 0.065859 | 0.014620 | 761.320 | 84.59 | 676.73 |
| C4 | −1191.592570 | 0.090762 | 0.018935 | 801.998 | 107.10 | 694.90 |
| C5 | −1489.530210 | 0.115841 | 0.022700 | 892.683 | 131.48 | 761.20 |
| C6 | −1787.409345 | 0.138252 | 0.027807 | 1111.278 | 160.23 | 951.05 |
| C7 | −2085.300259 | 0.161510 | 0.032395 | 1316.843 | 184.69 | 1132.15 |
| C8 | −2383.208447 | 0.185217 | 0.036890 | 1515.384 | 219.61 | 1295.77 |
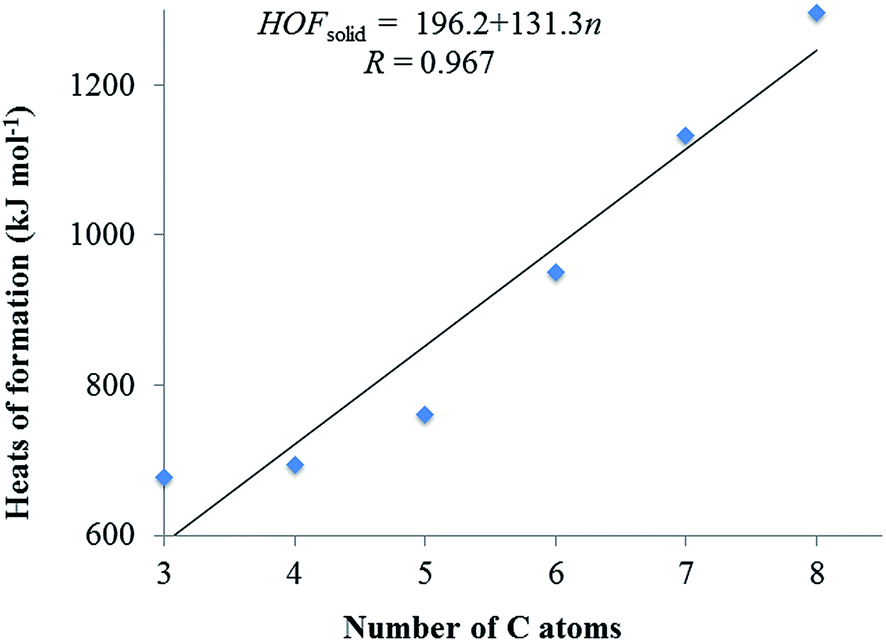 | ||
| Fig. 2 Heats of formation in solid phase and the relationship of C atoms for the designed compounds. | ||
3.2 Crystal structure and detonation properties
Packing motif strongly influences the electronic property and detonation performance of materials. The crystal packing of the title compounds was predicted by Dreiding force field which has been successfully applied for the condensed phase properties of hexanitroazobenzene derivatives.59 The approach is based on the generation of possible packing arrangements of the reasonable space groups including P21/c, P21, P212121, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Pbca, Pna21 and C2/c.60,61 Fig. 3 presents the crystal structures of the title compounds. The cell parameters and the crystal densities of the lowest pattern among the seven possible packing patterns are collected in Table 3. The crystal densities of the title compounds are in the range of 1.85–1.95 g cm−3, which generally measure up to crystal density criteria ρ ≈ 1.9 g cm−3 for HEDCs. Note that it was found in cyclic nitramines and polynitraminecubanes that introducing more nitramine groups in the molecule will increase the crystal density.58,62 However, the numbers of nitramine group in C3, C4 and C5 are less than that in C7 and C8, but the crystal density of the former are comparable to the latter. This is because C3, C4 and C5 respectively belong to the P21/c, P21 and P21/c space group, where the stacking patters are more compact. Meanwhile C7 and C8 belong to the P space group, where fewer molecules occupy one unit cell compared with those of C3, C4 and C5.
, Pbca, Pna21 and C2/c.60,61 Fig. 3 presents the crystal structures of the title compounds. The cell parameters and the crystal densities of the lowest pattern among the seven possible packing patterns are collected in Table 3. The crystal densities of the title compounds are in the range of 1.85–1.95 g cm−3, which generally measure up to crystal density criteria ρ ≈ 1.9 g cm−3 for HEDCs. Note that it was found in cyclic nitramines and polynitraminecubanes that introducing more nitramine groups in the molecule will increase the crystal density.58,62 However, the numbers of nitramine group in C3, C4 and C5 are less than that in C7 and C8, but the crystal density of the former are comparable to the latter. This is because C3, C4 and C5 respectively belong to the P21/c, P21 and P21/c space group, where the stacking patters are more compact. Meanwhile C7 and C8 belong to the P space group, where fewer molecules occupy one unit cell compared with those of C3, C4 and C5.
 | ||
| Fig. 3 Crystal structures of the title compounds. | ||
| Compd | Space group | Z | ρ (g cm−3) | a (Å) | b (Å) | c (Å) | α | β | γ |
|---|---|---|---|---|---|---|---|---|---|
| C3 | P21/c | 4 | 1.90 | 10.57 | 7.973 | 13.767 | 90 | 139.39 | 90 |
| C4 | P21 | 2 | 1.93 | 6.677 | 9.655 | 11.782 | 90 | 139.29 | 90 |
| C5 | P21/c | 4 | 1.95 | 10.891 | 23.938 | 16.733 | 90 | 163.68 | 90 |
| C6 | P |
2 | 1.85 | 7.565 | 7.564 | 14.765 | 105.1 | 97.73 | 103.27 |
| C7 | P |
2 | 1.90 | 8.951 | 14.309 | 7.479 | 71.84 | 77.42 | 79.58 |
| C8 | P |
2 | 1.94 | 8.656 | 9.036 | 15.016 | 83.73 | 57.76 | 87.07 |
Detonation velocity and detonation pressure, two important parameters to evaluate the detonation properties of energetic compounds, are calculated using the Kamlet–Jacobs empirical equations. Table 4 presents the detonation properties of the title compounds as well as RDX and HMX for comparison. It is clear that C3, C4, C5, C7, and C8 have better detonation properties than HMX. The detonation properties of C6 (D = 9.18 km s−1, P = 38.03 GPa) are higher than that of RDX (D = 8.75 km s−1, P = 34.00 GPa)63 and very close to that of HMX (D = 9.10 km s−1, P = 39.00 GPa).63 In addition, C5 has the largest crystal density (ρ = 1.95 g cm−3), but does not show the best explosive performances among the investigated molecules. However, C3, which owns the largest heat of detonation (Q = 2054.31 kJ g−1), shows the best detonation performance (D = 9.63 km s−1, P = 42.48 GPa) even though it does not possess the largest crystal density in all compounds. The result indicates that the heat of detonation is another very important factor in determination of the detonation performance besides the crystal density. In order to clarify the impacts of ρ and Q on detonation performance, the crystal densities, heats of detonation, detonation velocities, and detonation pressure of the designed compounds versus the ring size are plotted (see Fig. S1 in ESI†). Obviously, the detonation properties of C5, C6, C7 and C8 are predominantly controlled by their ρ, but the contribution of Q to detonation performance is large for C3 and C4.
| Compd | Q (kJ g−1) | ρ (g cm−3) | D (km s−1) | P (GPa) |
|---|---|---|---|---|
| C3 | 2054.31 | 1.90 | 9.63 | 42.48 |
| C4 | 1882.27 | 1.93 | 9.53 | 41.96 |
| C5 | 1811.12 | 1.95 | 9.51 | 42.01 |
| C6 | 1831.81 | 1.85 | 9.18 | 38.03 |
| C7 | 1842.56 | 1.90 | 9.37 | 40.23 |
| C8 | 1843.36 | 1.94 | 9.51 | 41.95 |
| RDX | 1591.03 | 1.81 | 8.75 | 34.00 |
| HMX | 1633.90 | 1.90 | 9.10 | 39.00 |
3.3 Explosive power and power index
Heat and gases are released in an explosive reaction. The volume of resulted gas provides information on how much work done by the explosive. Standard conditions should be set up in order to measure the volumes of generated gas, since the volume of gas is dependent on the temperature. The standard conditions (273 K, 1 atm) enable us to make comparisons between many explosives. A division of the value of total volume of gas produced upon detonation by the molecular weight provides an idea of how much gas is released per gram of explosive. Explosive power and power index can be calculated by eqn (7) and (8).52,64In order to illustrate the quantity and identity of the decomposition products, the Kistiakowsky and Wilson rule52 is used, that is, all the N atoms turn into N2; the O atoms react with H atoms to produce H2O at first; in case of O atoms are excessive, then C is converted to CO; after that, if some O atoms still remain, then CO is converted to CO2. The decomposition products of all designed compounds are listed (see Table S7 in ESI†). Note that the detonation products of the title compounds are N2 and CO2, which are environmental friendly compared with those containing CO, NxOy and HF released by other energetic materials such as HMX, 1,1′-methylenedioxy bis(2-fluoro-2,2-dinitroethane), and 2,2,2-trifluoro-1-nitoaminoethane.65
The explosive power index, which results from the explosive power of the investigated compound compared with that of a standard explosive, is an important parameter for energetic materials. The power index values of the title compounds and the reference molecule picric acid (PAc) and two classic energetic compounds 1-methyl-2,4,6-trinitrobenzene (TNT) and 3-nitro-1,2,4-triazol-5-one (NTO)66 are presented in Table 5. The results show that the power indexes of all designed compounds are between 98% and 111%, which are higher than that (87%) of NTO. The power index of C4, C5, C6, C7 and C8 are around 100% and very close to each other. The power index (111%) of C3 is the largest, which is even a litter better than that (110%) of TNT. In view of the detonation velocity, detonation pressure and explosive power index, we can conclude that all the designed N–NO2 substituted cycloalkanes derivatives exhibit impressive detonation performance. Among them, C3 might be the most promising candidate for HEDCs.
| Compd | Q (kJ g−1) | V (dm3 g−1) | QV | Power index% |
|---|---|---|---|---|
| C3 | 2054.31 | 0.622 | 1277.78 | 111 |
| C4 | 1882.27 | 0.622 | 1170.77 | 102 |
| C5 | 1811.12 | 0.622 | 1126.51 | 98 |
| C6 | 1831.81 | 0.622 | 1139.39 | 99 |
| C7 | 1842.56 | 0.622 | 1146.07 | 100 |
| C8 | 1843.36 | 0.622 | 1146.57 | 100 |
| Picric acid | 1379.07 | 0.831 | 1146.09 | 100 |
| TNT | 110 | |||
| NTO | 87 |
3.4 Thermal stability and sensitivity
To guarantee safe handling processes and enhance controllability of kinetic energy release, an ideal energetic compound must be thermally stable and insensitive toward destructive stimuli (impact/shock/friction/electrostatic discharge). The bond dissociation energy (BDE) can offer useful information about the stability of energetic compounds. Generally, the smaller the BDE for breaking a bond is, the weaker the bond is, and the easier the bond is broken. Nowadays, people have reached a consensus that the nitro group is often the primary cause of initiation reactivity of organic polynitro compounds.67 In addition, the bond order is another important parameter on describing the nature of various types of bond, where a smaller Wiberg bond index (WBI) usually indicates a weaker bond.68 Therefore, natural bond orbital (NBO) analyses were carried out to obtain the WBI of the designed compounds. Table 6 presents the bond lengths, the WBI, the h50 and the BDE of the N–NO2 bonds of the title compounds at B3LYP/6-31G** level. The bond lengths of the trigger N–NO2 lie in the range of 1.457–1.487 Å, and the WBI values are close to 0.9. There is good consistence between the bond length and the WBI value, where a shorter bond length corresponds to a large WBI value. The BDE values of the N–NO2 bond are between 100 and 120 kJ mol−1 for all compounds, where those of C3 and C6 are 102.39 and 105.35 kJ mol−1 while the other four are from 111 to 117 kJ mol−1. There does not exist clear correlation between the BDE and the size of the core ring. Importantly, all BDE values of the designed compounds are over the threshold of 80 kJ mol−1 (ref. 69) for HEDCs.| Compd | N–NO2 | C–C (ring) | h50 | ||||
|---|---|---|---|---|---|---|---|
| Bond length | WBI | BDE | Bond length | WBI | BDE | ||
| C3 | 1.471 | 0.8724 | 102.39 | 1.457 | 1.0112 | 104.25 | 59 |
| C4 | 1.468 | 0.8759 | 111.27 | 1.506 | 0.988 | 159.77 | 45 |
| C5 | 1.457 | 0.8884 | 116.73 | 1.495 | 1.0089 | 180.51 | 26 |
| C6 | 1.472 | 0.8784 | 105.35 | 1.516 | 0.9586 | 168.73 | 38 |
| C7 | 1.487 | 0.8667 | 112.26 | 1.528 | 0.9493 | 164.74 | 17 |
| C8 | 1.482 | 0.871 | 112.37 | 1.522 | 0.9498 | 167.41 | 17 |
Apart from BDE, the impact sensitivity (h50) has also been computed to evaluate the stability of the title compounds in this work. h50 is the height at which the sample is impacted by a given mass and there is 50% probability of causing an explosion. The greater is h50, the less sensitive to impact is a compound. h50 is frequently used to judge whether energetic compounds are sensitive or insensitive to external impact.70,71 The oxygen balance (OB100), and QNO2 are presented (see Table S8 in ESI†). Obviously, OB100 of the title compounds is equal due to the identical ratio of C, N and O. The h50 values of the title compounds lie in the range of 17–59 cm with the order of C3 > C4 > C6 > C5 > C7C8. These results show that the sensitivity upsurges as the number of N–NO2 groups increases. Moreover, we can see the order of h50 is not exactly consistent with that of BDE. For example, the BDE of C7 (112.26 kJ mol−1) is higher than that of C3 (102.39 kJ mol−1), but h50 of C7 (17 cm) is lower than that of C3 (59 cm). This supports the viewpoint of Politzer that any correlation between bond strength and impact sensitivity is not general, but limited within certain classes of molecules.68 The h50 values of C3, C4 and C6 are between 38 cm and 59 cm, which are higher than that of RDX (26 cm), HMX (29 cm), and CL-20 (12 cm). The sensitivity of C5 is very close to that of HMX and RDX. Most importantly, all the title compounds are less sensitive than CL-20.
3.5 Discussion
TheN–NO2 group is an important explosophore that is present in many energetic compounds. In experiment, quite a few compounds containing N–NO2 group have been successfully synthesized, for example 6-nitroimino-1,3,5-trinitro-1,3,5-triazacyclohexane derivatives,72 2-nitroimino-5-nitrohexahydroinitro-1,3,5-triazine,73 2-nitrimino-5-nitrohexahydro-1,3,5-triazine,74 nitroimino-tetrazole derivatives,1,75,76 3-methyl-4-nitroimino-tetrahydro-1,3,5-oxadiazine,77 neonicotinoid compounds,78 nitroguanidines.79 In this light, it is reasonable to conjecture the cycloalkane derivatives designed by our theoretical work can be synthesized in practice.
One important issue that should be pointed out is that, although the nitro group is often the primary cause of initiation, the C–C bonds of title molecules may break as the initial step, too. To comprehensively study the stability of title molecules, the ring breaking processes were simulated to compare with the N–NO2 cleavage routes. The C–C bond lengths and the corresponding WBI and BDE values are collected in Table 6. For compounds C4–C8, the C–C bonds exhibit 49–64 kJ mol−1 higher BDE values than those of the corresponding N–NO2 bonds, indicating that the N–NO2 bond breaking is much more possible to be the initiation decomposition step in these compounds. As for C3, the BDE value of the C–C bond is about 2 kJ mol−1 larger than that of the N–N bond, suggesting that the latter is slightly prior to the former as the cause of initiation. In short, the breaking of N–N bonds is the preferred initial step in the title compounds.
Note that the 6-31G* basis set was employed in the original paper of eqn (4), but here we use a bigger basis set 6-31G**, which has proved to be a good choice to the get the HOF values that are very close to the experimental data.80 Similarly, in the original paper of eqn (11), the calculations were carried out by using semi-empirical method on MOPAC program.56 We performed the calculations at B3LYP/6-31G** level because this accurate method has been successfully applied to get the h50 value based on eqn (11).81–83
In addition, the above results were obtained at the DFT-B3LYP level of theory. In order to better assess our conclusions, the performance parameters including heat of formation, detonation properties, and impact sensitivity index values of C3(N–NO2)3 and C6(N–NO2)6 were recalculated at MP2/6-31G** level and summarized in Table S9.† In general, the trends of these performance parameters at MP2/6-31G** level are consistent with those at DFT-B3LYP level. This demonstrates the robustness of our conclusions.
4 Conclusions
In this work, we theoretically designed a set of N–NO2 substituted cycloalkanes derivatives Cm(N–NO2)m for energetic materials. The geometric and crystal structures, gas-phase and solid-phase HOFs, detonation performance, and impact sensitivity of the designed compounds were systematically at B3LYP/6-31 G** level. The results show that the designed compounds have large crystal density (1.85–1.95 g cm−3) and possess large positive HOFs that increase with the growing number of the N–NO2 groups. Importantly, the title compounds exhibit remarkable detonation performance compared with the classic explosives RDX, HMX, CL-20. Furthermore, the bond dissociation energies of larger than 100 kJ mol−1 illustrate the designed molecules relatively stable. The impact sensitivity index h50 of 17–59 cm reveals the title compounds are less sensitive than CL-20. In conclusion, the designed molecules are expected to be promising for energetic materials with high detonation performance and low impact sensitivity.Acknowledgements
This work is financially supported by the Major State Basic Research Development Programs of China (2011CBA00701), the National Natural Science Foundation of China (21473010, 20933001). This work is also supported by the opening project of State Key Laboratory of Explosion Science and Technology (Beijing Institute of Technology) (ZDKT12-03).References
- R. Wang, H. Xu, Y. Guo, R. Sa and J. M. Shreeve, J. Am. Chem. Soc., 2010, 132, 11904–11905 CrossRef CAS PubMed.
- V. Thottempudi, H. Gao and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 6464–6471 CrossRef CAS PubMed.
- X. Jin, B. Hu, Z. Liu and C. Lv, RSC Adv., 2014, 4, 23898–23903 RSC.
- P. F. Pagoria, G. S. Lee, A. R. Mitchell and R. D. Schmidt, Thermochim. Acta, 2002, 384, 187–204 CrossRef CAS.
- Q. H. Zhang and J. M. Shreeve, Angew. Chem., Int. Ed., 2014, 53, 2540–2542 CrossRef CAS PubMed.
- F. Wang, G. X. Wang, H. C. Du, J. Y. Zhang and X. D. Gong, J. Phys. Chem. A, 2011, 115, 13858–13864 CrossRef CAS PubMed.
- L. Qiu, H. M. Xiao, X. D. Gong, X. H. Ju and W. H. Zhu, J. Phys. Chem. A, 2006, 110, 3797–3807 CrossRef CAS PubMed.
- F. Wang, H. C. Du, J. Y. Zhang and X. D. Gong, J. Phys. Chem. A, 2011, 115, 11788–11795 CrossRef CAS PubMed.
- Y. Pan, J. S. Li, B. B. Cheng, W. H. Zhu and H. M. Xiao, Comput. Theor. Chem., 2012, 992, 110–119 CrossRef CAS PubMed.
- W. J. Chi, X. Y. Wang, B. T. Li and H. S. Wu, J. Mol. Model., 2012, 18, 4217–4223 CrossRef CAS PubMed.
- L. B. Munday, P. W. Chung, B. M. Rice and S. D. Solares, J. Phys. Chem. B, 2011, 115, 4378–4386 CrossRef CAS PubMed.
- M. H. Keshavarz, J. Hazard. Mater., 2011, 190, 330–344 CrossRef CAS PubMed.
- Q. Wu, W. H. Zhu and H. M. Xiao, RSC Adv., 2014, 4, 3789–3797 RSC.
- A. Sikder and N. Sikder, J. Hazard. Mater., 2004, 112, 1–15 CrossRef CAS PubMed.
- H. J. Singh, M. K. Upadhyay and S. K. Sengupta, J. Mol. Model., 2014, 20, 1–10 CAS.
- X. W. Fan and X. H. Ju, J. Comput. Chem., 2008, 29, 505–513 CrossRef CAS PubMed.
- L. Qiu, X. D. Gong, G. X. Wang, J. Zheng and H. M. Xiao, J. Phys. Chem. A, 2009, 113, 2607–2614 CrossRef CAS PubMed.
- M. Zamani and M. H. Keshavarz, Comput. Mater. Sci., 2015, 97, 295–303 CrossRef CAS PubMed.
- T. T. Vo, J. Zhang, D. A. Parrish, B. Twamley and J. M. Shreeve, J. Am. Chem. Soc., 2013, 135, 11787–11790 CrossRef CAS PubMed.
- J. Zhang and J. M. Shreeve, J. Am. Chem. Soc., 2014, 136, 4437–4445 CrossRef CAS PubMed.
- W. J. Chi and Z. S. Li, RSC Adv., 2015, 5, 7766–7772 RSC.
- Q. Wu, W. H. Zhu and H. M. Xiao, J. Mater. Chem. A, 2014, 2, 13006–13015 CAS.
- J. G. Zhang, P. He, K. Wang, X. Yin, X. Jin and T. L. Zhang, Phys. Chem. Chem. Phys., 2015, 17, 5840–5848 RSC.
- H. Lin, S. G. Zhu, L. Zhang, X. H. Peng, P. Y. Chen and H. Z. Li, Int. J. Quantum Chem., 2013, 113, 1591–1599 CrossRef CAS PubMed.
- J. F. Liebman and A. Greenberg, Chem. Rev., 1976, 76, 311–365 CrossRef CAS.
- J. A. Boatz, M. S. Gordon and R. L. Hilderbrandt, J. Am. Chem. Soc., 1988, 110, 352–358 CrossRef CAS.
- S. Staudt, E. Burda, C. Giese, C. A. Müller, J. Marienhagen, U. Schwaneberg, W. Hummel, K. Drauz and H. Gröger, Angew. Chem., Int. Ed., 2013, 52, 2359–2363 CrossRef CAS PubMed.
- A. L. Pitts, A. Wriglesworth, X. Z. Sun, J. A. Calladine, S. D. Zaric, M. W. George and M. B. Hall, J. Am. Chem. Soc., 2014, 136, 8614–8625 CrossRef CAS PubMed.
- W. J. Chi, B. T. Li and H. S. Wu, Struct. Chem., 2013, 24, 375–381 CrossRef CAS PubMed.
- X. L. Zhang and X. D. Gong, J. Mol. Model., 2015, 20, 26–36 CrossRef PubMed.
- W. J. Chi, L. L. Li, B. T. Li and H. S. Wu, Struct. Chem., 2012, 23, 1837–1841 CrossRef CAS.
- H. M. Xiao, X. J. Xu and L. Qiu, Theoretical Design of High Energy and Density Materials, Science Press, Beijing, 2008 Search PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218–8224 CrossRef CAS PubMed.
- R. Bauernschmitt and R. Ahlrichs, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439–4449 CrossRef CAS PubMed.
- T. Wei, W. H. Zhu, J. J. Zhang and H. M. Xiao, J. Hazard. Mater., 2010, 179, 581–590 CrossRef CAS PubMed.
- G. X. Wang, X. D. Gong, Y. Liu and H. M. Xiao, Int. J. Quantum Chem., 2010, 110, 1691–1701 CAS.
- T. Wei, W. H. Zhu, X. W. Zhang, Y. F. Li and H. M. Xiao, J. Phys. Chem. A, 2009, 113, 9404–9412 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. J. Frisch, J. A. Pople and J. S. Binkley, J. Chem. Phys., 1984, 80, 3265–3269 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. Petersson, Gaussian 09 D.01, Inc., Wallingford, CT, 2010 Search PubMed.
- H. M. Xiao and Z. X. Chen, The modern theory for tetrazole chemistry, Science Press, Beijing, 2000 Search PubMed.
- X. H. Ju, Y. M. Li and H. M. Xiao, J. Phys. Chem. A, 2005, 109, 934–938 CrossRef CAS PubMed.
- P. W. Atkins, Physical chemistry, Oxford University Press, Oxford, 1982 Search PubMed.
- P. Politzer, J. S. Murray, M. E. Grice, M. Desalvo and E. Miller, Mol. Phys., 1997, 91, 923–928 CrossRef CAS.
- P. Politzer and J. S. Murray, Cent. Eur. J. Energ. Mater., 2011, 8, 209–220 CAS.
- E. F. Byrd and B. M. Rice, J. Phys. Chem. A, 2006, 110, 1005–1013 CrossRef CAS PubMed.
- F. A. Bulat, A. Toro-Labbé, T. Brinck, J. S. Murray and P. Politzer, J. Mol. Model., 2010, 16, 1679–1691 CrossRef CAS PubMed.
- M. J. Kamlet and S. Jacobs, J. Chem. Phys., 1968, 48, 23–35 CrossRef CAS PubMed.
- Accelrys Software Materials Studio, Accelrys Software, Inc., San Diego, 2007 Search PubMed.
- S. L. Mayo, B. D. Olafson and W. A. Goddard, J. Phys. Chem., 1990, 94, 8897–8909 CrossRef CAS.
- J. Akhavan, The chemistry of explosives, Royal Society of Chemistry, 2011 Search PubMed.
- A. Martin and H. Yallop, J. Appl. Chem., 1959, 9, 310–315 CrossRef CAS PubMed.
- B. M. Rice and E. F. Byrd, J. Mater. Res., 2006, 21, 2444–2452 CrossRef CAS.
- Z. Yu and E. R. Bernstein, J. Phys. Chem. A, 2013, 117, 10889–10902 CrossRef CAS PubMed.
- C. Cao and S. Gao, J. Phys. Chem. B, 2007, 111, 12399–12402 CrossRef CAS PubMed.
- http://webbook.nist.gov/chemistry/form-ser.html.
- G. Zhao and M. Lu, J. Phys. Org. Chem., 2014, 27, 10–17 CrossRef CAS PubMed.
- Y. Liu, X. D. Gong, L. J. Wang, G. X. Wang and H. M. Xiao, J. Phys. Chem. A, 2011, 115, 1754–1762 CrossRef CAS PubMed.
- N. Y. Chernikova, V. Bel'skii and P. Zorkii, J. Struct. Chem., 1990, 31, 661–666 CrossRef.
- A. Wilson, Acta Crystallogr., Sect. A: Found. Crystallogr., 1988, 44, 715–724 CrossRef.
- W. J. Chi, L. L. Li, B. T. Li and H. S. Wu, J. Mol. Model., 2012, 19, 571–580 CrossRef PubMed.
- M. Talawar, R. Sivabalan, T. Mukundan, H. Muthurajan, A. Sikder, B. Gandhe and A. S. Rao, J. Hazard. Mater., 2009, 161, 589–607 CrossRef CAS PubMed.
- L. Tuerker and S. Variş, J. Energ. Mater., 2013, 31, 203–216 CrossRef.
- M. H. Keshavarz and H. R. Pouretedal, J. Chin. Inst. Eng., 2006, 29, 145–151 CrossRef CAS.
- L. Türker and S. Variş, Z. Anorg. Allg. Chem., 2014, 640, 208–212 CrossRef PubMed.
- X. H. Jin, B. C. Hu, W. Lu, S. J. Gao, Z. L. Liu and C. X. Lv, RSC Adv., 2014, 4, 6471–6477 RSC.
- J. S. Murray, M. C. Concha and P. Politzer, Mol. Phys., 2009, 107, 89–97 CrossRef CAS.
- G. Chung, M. W. Schmidt and M. S. Gordon, J. Phys. Chem. A, 2000, 104, 5647–5650 CrossRef CAS.
- B. M. Rice and J. J. Hare, J. Phys. Chem. A, 2002, 106, 1770–1783 CrossRef CAS.
- P. Politzer and J. S. Murray, J. Mol. Model., 2014, 20, 2223–2230 CrossRef PubMed.
- Y. Yongzhong, S. Zhuang, D. Baoru and C. Fubo, Propellants, Explos., Pyrotech., 1989, 14, 150–152 CrossRef PubMed.
- M. D. Cliff, I. J. Dagley, R. P. Parker and G. Walker, Propellants, Explos., Pyrotech., 1998, 23, 179–181 CrossRef CAS.
- R. W. Millar, J. Hamid, R. Endsor, P. F. Swinton and J. Cooper, Propellants, Explos., Pyrotech., 2008, 33, 66–72 CrossRef CAS PubMed.
- Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2009, 48, 564–567 CrossRef CAS PubMed.
- Y. H. Joo and J. M. Shreeve, Angew. Chem., Int. Ed., 2010, 49, 7320–7323 CrossRef CAS PubMed.
- W. Y. Qu, D. M. She, J. Zhao, D. J. Lin, Q. L. Huang and F. M. Li, Synth. Commun., 2012, 42, 1950–1958 CrossRef CAS.
- Y. Zhao, G. Wang, Y. Li, S. Wang and Z. Li, Chin. J. Chem., 2010, 28, 475–479 CrossRef CAS PubMed.
- P. Maienfisch, H. Huerlimann and J. Haettenschwiler, Tetrahedron Lett., 2000, 41, 7187–7191 CrossRef CAS.
- H. Liu, F. Wang, G. X. Wang and X. D. Gong, J. Comput. Chem., 2012, 33, 1790–1796 CrossRef CAS PubMed.
- Q. Cao, J. Mol. Model., 2013, 19, 2205–2210 CrossRef CAS PubMed.
- J. Bai, W. J. Chi, L. L. Li, T. Yan, X. E. Wen, B. T. Li, H. S. Wu and F. L. Ma, Cent. Eur. J. Energ. Mater., 2013, 10, 467–480 CAS.
- P. Srinivasan and P. Kumaradhas, Cent. Eur. J. Energ. Mater., 2013, 10, 53–68 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04509f |
| This journal is © The Royal Society of Chemistry 2015 |