
Preparation of high-quality graphene with a large-size by sonication-free liquid-phase exfoliation of graphite with a new mechanism
Rui Zhanga,
Baochang Zhangb and
Shuqing Sun*a
aDepartment of Chemistry, College of Science, Tianjin University, Tianjin, 300072, China. E-mail: sunshuqing@tju.edu.cn
bCollege of Chemistry and Life Sciences, Changchun University of Technology, Chang Chun, 130012, China
First published on 11th May 2015
Abstract
In this paper, we report on the successful preparation of high-quality graphene sheets with a large-size in N,N-dimethylformamide (DMF) solvent by sonication-free liquid-phase exfoliation (LPE) of graphite assisted with urea as the precursor of the intercalating agent melamine. Through the characterizations of the graphene sheets obtained with transmission electron microscopy (TEM), atomic force microscopy (AFM), Raman spectroscopy and XPS, these graphene sheets were found to be high-quality, defect-free, single or-few-layer with areas in the range of 100–1000 μm2. The influences of reflux time and the mass ratio of urea to graphite on the graphene concentration (CG) in the resulting dispersions were investigated. The graphene dispersions were fabricated into flexible films that show comparable conductivity. The exfoliation mechanism of graphite in our new sonication-free LPE method is also discussed.
1. Introduction
In the last decade graphene, a two-dimensional one-atom-thick honeycomb lattice carbon network, has emerged as an exciting material possessing unique properties such as outstanding electronic,1 thermal,2 mechanical3 and optical properties.4 Therefore, it has attracted increasing attention in various potential applications including electronics and photonics,5–7 energy conversion and storage,7,8 polymer nanocomposites,9 and catalysis.10 Until now, several methods have been developed for preparing single-layer or few-layer graphene sheets, such as reduction of graphene oxide,11 mechanical exfoliation,12 chemical vapor deposition (CVD)13 and chemical synthesis.14 Clearly, each method has its advantages and disadvantages: reduction of graphene oxide suffers from drawbacks related to incomplete retention of the sp2 carbon network, and mechanical exfoliation was shown to have a low yield, and graphene films deposited by CVD are difficult to transfer from the substrate as well as the sizes of graphene generated by chemical synthesis being less than the ones by other methods.For these reasons, large-scale preparation of high-quality graphene with the fewer defects and larger size remains a challenge. Consequently, an alternative method has been explored for this purpose by LPE of pristine graphite in some particular organic solvents such as DMF, N-methylpyrrolidone and o-dichlorobenzene.15–17 These solvents have the surface energy matching well with that of graphene, so graphite can be exfoliated in these solvents with the aid of ultrasound process, which splits the graphite crystallites into single-layer or few-layer graphene sheets. LPE method has the advantage of giving stable dispersions of graphene sheets.16 However, the sonication step has the major effect on the size distribution of the graphene sheets and the sizes of the graphene sheets decrease with increasing ultrasound time. For example, with the help of long-time ultrasonic treatment of natural graphite in the solvent of N-methyl-pyrrolidone.18 Coleman's group achieved graphene sheets with average length and width of 1 μm and 300 nm, respectively. Hamilton et al. reported that the graphene sheets with lateral size in the range of 100–500 nm were obtained by sonicating graphite in ortho-dichlorobenzene.19 Xu et al.20 prepared few-layer graphene sheets with the lateral dimension ranging from 200 to 500 nm via sonicating graphite in the solvent with the assistance of hyperbranched polyethylene. LPE of graphite generally results in sheets with lateral size of one micrometer or less, hence being too small for many applications.
Large-sized graphene exhibits many excellent properties such as ultrahigh electrical, thermal conductivity and outstanding mechanical21–23 properties, resulting in potential applications including electrical improvement of devices and mechanical reinforcement of composites. To enhance the size of graphene, Coleman and co-workers24 reported a method to separate an existing dispersion with different centrifugation rates, resulted in a range of dispersions with the mean sheet length varying from 1 μm for the highest centrifugation rate to 3.5 μm for the lowest centrifugation rate. The CVD method has been most widely employed to grow large area graphene sheets on metal surfaces.25,26 However, this method is an energy intensive process that might be too expensive for many applications.27
Therefore, this work is specifically aimed at developing sonication-free LPE method in which urea was used as the precursor of intercalating agent melamine. By using this method, the high-quality and large-size graphene sheets with single-layer or few-layer were produced. The structure, morphology and size distribution of the as-exfoliated graphene sheets have been characterized by a variety of methods, including transmission electron microscopy (TEM), atomic force microscopy (AFM), scanning electron microscopy (SEM) and Raman spectroscopy. The exfoliation mechanism of graphite in our new LPE process is proposed and discussed in this work.
2. Experimental
Flake graphite powder (80 mesh, i.e., 178 μm) was purchased from Qingda Tianheda Graphite Co.,Ltd. Analytical grade urea and DMF were obtained from Aladdin and used as received.The exfoliation of graphite assisted with urea to prepare graphene sheets was undertaken by using the reflux of the mixture in DMF and following centrifugation treatment and the extraction of the graphene sheets. In a typical process, 1.2 g of graphite powder and 10.8 g of urea were dispersed in 120 mL DMF by magnetic stirring at room temperature for 30 min in a three-necked flask and then the suspension was allowed to stand for ca. 1 week to allow the graphite to be wetted fully by the DMF and urea molecules. Following the temperature was increased to boiling point of DMF (154 °C) and kept reflux for 12 h to allow the melamine to be formed by the urea and the graphite to be intercalated and exfoliated by the melamine. The resulting mixture was subsequently centrifuged at 4000 rpm for 20 min and the supernatant containing graphene sheets was then collected.
For the extraction of the graphene sheets, the as-prepared dispersion in DMF was filtered onto a 20 nm alumina membrane and thoroughly washed with hot water. It is expected that unreacted urea and formed melamine were removed from dispersion because urea and melamine can be dissolved in hot water. For the characterization of the graphene sheets, they were redispersed into tetrahydrofuran (THF) solvent.
TEM samples were prepared by depositing a few drops of the dispersion onto holey carbon grids (400 mesh). TEM images and selected area electron diffraction were captured on a Jeol JEM-2100F transmission electron microscope operated at 100 keV. Powder X-ray diffraction (PXRD) data was collected on a Rigaku D/max-2550 diffractometer with CuKα radiation (λ = 1.5418 Å). The morphologies and sizes of the graphene sheets were investigated using a Hitachi S-4200 field emission scanning electron microscope and a scanning electron microscope (SEM, XL-30, FEG). AFM (Scanning Probe Microscope-NanoScope, Digital Instruments) was used to characterize the layered structure and thickness of graphene sheets. Gas sorption experiments were performed at 77 K and 0–760 Torr on a Micromeritics ASAP 2020 instrument. UV-vis spectroscopic analysis was performed on an Ultraspec 2100 Pro UV-vis spectrophotometer. The quality of graphite and graphene sheets were analyzed by micro-Raman spectroscopy in confocal configuration (NT-MDT NTEGRA Spectra, with 532 nm laser). X-ray photoelectron spectroscopy (XPS, Axis Ultra DLD) was used to characterize the elemental compositions and the assignments of the carbon peaks of graphene sheets. Electrical conductivity was measured with a Four Probes Tech RTS-9 four-point-probe conductivity tester (Guangzhou, China) at ambient temperature. The samples were pressed at 5 MPa for electrical conductivity measurements. The TG and DSC curves of the graphene were determined directly by filling graphene powder into a thermogravimetric analysis (TGA) sample pan. Before measurement, the sample was dried in vacuum oven at 180 °C for 5 h and was subsequently heated to 800 °C at 10 °C min−1 in N2 atmosphere on a TA Q50 thermogravimetric analyzer.
3. Results and discussion
3.1 Preparation and quality of graphene sheets
The graphene sheets were prepared by using a new sonication-free LPE method. The structures and morphologies of exfoliated graphene sheets were characterized with the use of TEM, AFM, PXRD and Raman spectroscopy. As shown in Fig. 1a, TEM image of exfoliated graphene reveals that the large-size graphene sheet is transparent under the electron beam, suggesting a single-layer graphene sheet was exfoliated from graphite. Careful examination on the edge structure by tilting the sample proves the single-layer character of the graphene sheet. In addition, the graphene sheet is shown to be large-size and it has lateral dimensions of over 10 μm. The electron diffraction (ED) pattern, as shown in Fig. 1b, demonstrates the typical six fold symmetry characteristic diffraction for graphene with an ordered well-crystallized graphene structure. It corresponds to the circle-marked region of the graphene sheet (Fig. 1a), in which spots (0![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 10) and (1010) are more intense than spots (1
10) and (1010) are more intense than spots (1![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 10) and (2110). This finding reconfirms the single-layer feature of the graphene sheet.28 Fig. 1c shows another typical image of a single-layer graphene sheet. It is noted that the edge of the graphene sheet is slightly folded, revealing the graphene sheet exfoliated with flexibility. Fig. 1d shows a HRTEM image from the square-marked area (Fig. 1c). It shows the presence of a highly ordered hexagonal lattice with a measured spacing of 0.25 nm, which corresponds to the centre-to-centre distance of two graphene unit cells. The flat surface of our graphene is in contrast to the wrinkled surface of graphene oxide11 and this difference may suggest that our graphene has fewer topological defects. A bi-layer graphene sheet on the basis of its edge structure also appears in TEM image (Fig. 1e). As a comparison, the mixture was treated by ultrasonic LPE process for same time at 35 °C, resulting in few-lever graphene with smaller sizes (Fig. 1f).
10) and (2110). This finding reconfirms the single-layer feature of the graphene sheet.28 Fig. 1c shows another typical image of a single-layer graphene sheet. It is noted that the edge of the graphene sheet is slightly folded, revealing the graphene sheet exfoliated with flexibility. Fig. 1d shows a HRTEM image from the square-marked area (Fig. 1c). It shows the presence of a highly ordered hexagonal lattice with a measured spacing of 0.25 nm, which corresponds to the centre-to-centre distance of two graphene unit cells. The flat surface of our graphene is in contrast to the wrinkled surface of graphene oxide11 and this difference may suggest that our graphene has fewer topological defects. A bi-layer graphene sheet on the basis of its edge structure also appears in TEM image (Fig. 1e). As a comparison, the mixture was treated by ultrasonic LPE process for same time at 35 °C, resulting in few-lever graphene with smaller sizes (Fig. 1f).
 | ||
| Fig. 1 (a) TEM imagine of single-layer graphene sheet with the lateral dimensions of over 10 μm; (b) the ED pattern corresponding to the circle-marked area in image (a); (c) TEM image of another single-layer graphene sheet; (d) a magnified image obtained from the square-marked area of image (c); (e) TEM image of bi-layer graphene sheet. (a)–(e) Images taken from samples by sonication-free LPE method. (f) TEM image of few layer graphene sheets with less sizes and taken from sample by ultrasound LPE process. | ||
 | ||
| Fig. 2 (a) PXRD spectrum of the flake graphite and (b) PXRD spectrum of the graphene sheets. The inset represents (b) enlarged. | ||
PXRD analysis was performed on the graphene sheets and flake graphite powder. Their PXRD patterns are shown in Fig. 2. The spectrum of the graphite reflects one prominent peak at 26.6°, corresponding to the (002) planes of graphite, while for the pattern of the graphene sheets there is one superimposed reflection peak at 23.4°. It is expected that the carbon (002) peak at 26.6° (d = 0.334 nm) is relative to the ordered graphite crystals of flake graphite and 23.4° (d = 0.386) relates to a larger interlayer spacing of lattice-damaged crystals of the graphene sheets. Misalignments and defects in the carbon layer stacks with a larger carbon interlayer spacing were responsible for broadening of the XRD carbon peak (002). There is no other impurity peaks in the PXRD patterns of the graphene sheets, indicating that the purity of the graphene materials prepared is over 95%.
AFM was used to measure the morphology and number of layers of graphene sheets by measuring the height of the deposited sheets. AFM measurement was conducted on graphene dispersion in THF that was dropped onto silicon oxide substrate and then dried at room temperature. AFM analysis further supports the TEM results. Fig. 3 presents the higher-magnification AFM images for two individual sheets. From their corresponding height profiles (Fig. 3), they show uniform surfaces and their height was measured to be about 1.1 nm. Since on SiO2, a single-layer graphene sheet can appear to have a height of ∼1 nm, while on mica it amounts to ∼0.4 nm,29 thus the two sheets should be single-layer graphene. The AFM results also confirm that the graphene sheets with large areas can be obtained by our sonication-free LPE method.
 | ||
| Fig. 3 AFM-image of graphene sheets deposited on oxidized Si substrate and height profile along the line in image. | ||
In order to determine the quality of the prepared graphene sheets, Raman spectroscopic analysis was carried out by using a high resolution dispersive Raman microscope with an excitation laser of 532 nm. Fig. 4 compares the Raman spectra taken graphene sample and graphite powder. In first-order Raman spectrum of pristine graphite (Fig. 4a), a G band at 1571 cm−1, a 2D band at 2720 cm−1 and a very weak D band at 1343 cm−1 are observed. The G band is the result of first-order scattering of E2g mode of graphite and is related to the in-plane vibration of sp2 hybridized carbon–carbon bonds. It is common for pristine graphene not to have enough structural defects for the D and D′ peaks to be Raman active so that they can only be seen as very weak peaks. However, when graphene is affected by defects, D band at 1343 cm−1 and D′ band at 1609 cm−1 appear in Raman spectrum as shown in Fig. 4a. The intensity of D peak is attributed to the amount of disorder but does not relate to the number of graphene layers.30 The intensity ratio of D- and G-bands (ID/IG) is found to be 0.33 and 0.15 in the graphene sheets and pristine graphite, respectively. The ratio of 0.33 is smaller than typical values (0.35–0.7) reported in the literature for some high-quality graphene sheets obtained by LPE.31–34 It is thus concludes that the graphene sheets exfoliated by our new LPE method have high quality. The second-order Raman spectrum is very important because it allows qualitative identification of the thickness of the graphene sheets through the 2D peak shape. As shown in Fig. 4b, a distinct difference in the shape of the 2D-band is noted between the two spectra of the pristine graphite and graphene sheets. The pristine graphite presents an asymmetric and split 2D band consisting of two components of different intensity with the peak maximum position at 2720.5 and 2690 cm−1, respectively. The 2D-band in the graphene sample is a single and symmetrical peak with the peak maximum position moved to ca. 2686.1 cm−1, indicative of a very thin graphene sheet, probably a single-layer graphene sheet. Table 1 provides data for the Raman spectra carried out on multiple spots of graphene sheets, giving the average position of D, G, D′ and 2D peaks in the as prepared samples.
 | ||
| Fig. 4 Comparison of Raman spectra (excitation at 532 nm) for a graphene film deposited on an glass membrane and the pristine graphite. The graphene film was obtained by filtering a graphene dispersion in DMF onto an alumina membrane followed with washing and drying. | ||
| Spot | D (cm−1) | G (cm−1) | D′ (cm−1) | 2D (cm−1) |
|---|---|---|---|---|
| 1 | 1343.4 | 1571.4 | 1609.4 | 2686.1 |
| 2 | 1345.3 | 1573.6 | 1610.9 | 2690.4 |
| 3 | 1342.6 | 1571.5 | 1608.6 | 2687.5 |
| 4 | 1344.4 | 1572.7 | 1609.7 | 2691.2 |
| 5 | 1341.8 | 1570.4 | 1607.4 | 2688.5 |
| Average position | 1343.5 | 1571.9 | 1609.2 | 2688.7 |
Fig. 5a presents a typical SEM image of the graphene sheets by sonication-free LPE approaches, indicating the effective exfoliation of graphite flakes into many monolayer or few-layer graphene sheets with large sizes. For the statistical analysis, a histogram plotted as a function of size is obtained from the SEM image (Fig. 5a). The average area of the graphene sheets is about 400 μm2 by counting 42 sheets. As far as we know, there is not monolayer graphene prepared by LPE is larger than our monolayer graphene in size. Fig. 5b shows a histogram revealing the distribution of the areas for the graphene sheets. It is clearly that about 67% of the graphene sheets are in the range of 100–1000 μm2 and approximately 46% of the graphene sheets have sizes over 400 μm2, suggesting that it only may need to exfoliate mc-G on c-axis direction to acquire thin and large graphene sheets. This reduces energy consumption during the exfoliation and obtains large size and thin graphene sheets, bringing tremendous economic benefits. The resulting large-size graphene sheets are considered quite sufficient as precursors for many multifunctional applications such as conductive films, optoelectronic devices and graphene-polymer composites.
 | ||
| Fig. 5 (a) Typical SEM image of resultant graphene sheets deposited on a Si substrate through sonication-free LPE method. (b) Size distributions corresponding to the graphene sheets in image (a); (c) SEM image of the graphene film with smooth surface; (d) SEM image of the graphene film surface scratched by a small tip of tweezers to form a pleated film. | ||
Thin films consisting of the graphene sheets were deposited on silicon wafer using a drop casting. Fig. 5c shows the SEM image of the film surface and the film is relatively smooth without breaking and significant porosity. Fig. 5d displays the film surface scratched by a small tip of tweezers to form a pleated film, indicating that the film is thin and flexible. In order to measure the electrical conductivity of the graphene the circular membrane consisting of the graphene sheets was prepared by tableting machine. The conductivity of the graphene membrane was measured to be 7800 S m−1 with the four probe measurement technique. This value is closed compared to those reported in the literature for various graphene film prepared from dispersions obtained such as those obtained from graphene dispersions in NMP (6500–18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 S m−1)35,36 and those obtained from reduced GO (7000–10000 S m−1).37–39 In order to explore the surface area of the graphene material obtained, about 120 mg of the graphene material was activated at 100 °C for 8 h, and then gas sorption experiment was performed at 77 K and 0–760 Torr on a Micromeritics ASAP 2020 instrument. As evident by the reversible type I N2 sorption isotherms (see Fig. 6), an amount of N2 at 77 K (77 cm3 g−1 at 1 bar) was achieved. The estimated Langmuir surface area and BET surface area of the graphene material are 59.4 m2 g−1 and 42 m2 g−1, respectively. These values were less than the surface areas of the graphene materials in previous works,40 likely due to shrinking or overlapping of graphene sheets within the self-aggregation and drying process.
000 S m−1)35,36 and those obtained from reduced GO (7000–10000 S m−1).37–39 In order to explore the surface area of the graphene material obtained, about 120 mg of the graphene material was activated at 100 °C for 8 h, and then gas sorption experiment was performed at 77 K and 0–760 Torr on a Micromeritics ASAP 2020 instrument. As evident by the reversible type I N2 sorption isotherms (see Fig. 6), an amount of N2 at 77 K (77 cm3 g−1 at 1 bar) was achieved. The estimated Langmuir surface area and BET surface area of the graphene material are 59.4 m2 g−1 and 42 m2 g−1, respectively. These values were less than the surface areas of the graphene materials in previous works,40 likely due to shrinking or overlapping of graphene sheets within the self-aggregation and drying process.
 | ||
| Fig. 6 N2 adsorption isotherm for the graphene material at 77 K. Filled and open circles represent adsorption and desorption, respectively. | ||
3.2 Exfoliation mechanism
In order to investigate the exfoliation mechanism of graphite in our method, another control experiment was done, that is, same exfoliation process without urea. It was found that the yield of graphene exfoliated was different. The dark stable graphene dispersion was obtained in the DMF containing urea, while the resulting dispersion in DMF alone is grey. Therefore, the graphene was efficiently produced only when urea was present in a DMF solution by using sonication-free LPE method. We propose that there were two factors that resulted in exfoliation of graphene sheets from the surface of graphite flakes. Firstly, DMF molecular adsorption on the surface of graphite can weaken van der Waals interactions between graphite layers, which can facilitate the exfoliation of graphene sheets from graphite flakes.41,42 Furthermore, the exfoliation of graphite in DMF solvent could be assisted by forming intercalation agent melamine (Fig. 5). It is well known that urea can transfer into melamine at boiling point of DMF (154 °C) and melamine was an excellent intercalation agent in mill method for the preparation of single and few-levers graphene.43 As a result, intercalation effect of melamine was favourable to diminish van der Waals interactions between graphite layers, resulting in that graphene sheets can be exfoliated from graphite under sonication-free LPE. The corresponding equations and diagram (Fig. 7) are shown as follows:| 6CO(NH2)2 → C3H6N6 + 6NH3 + 3CO2 | (1) |
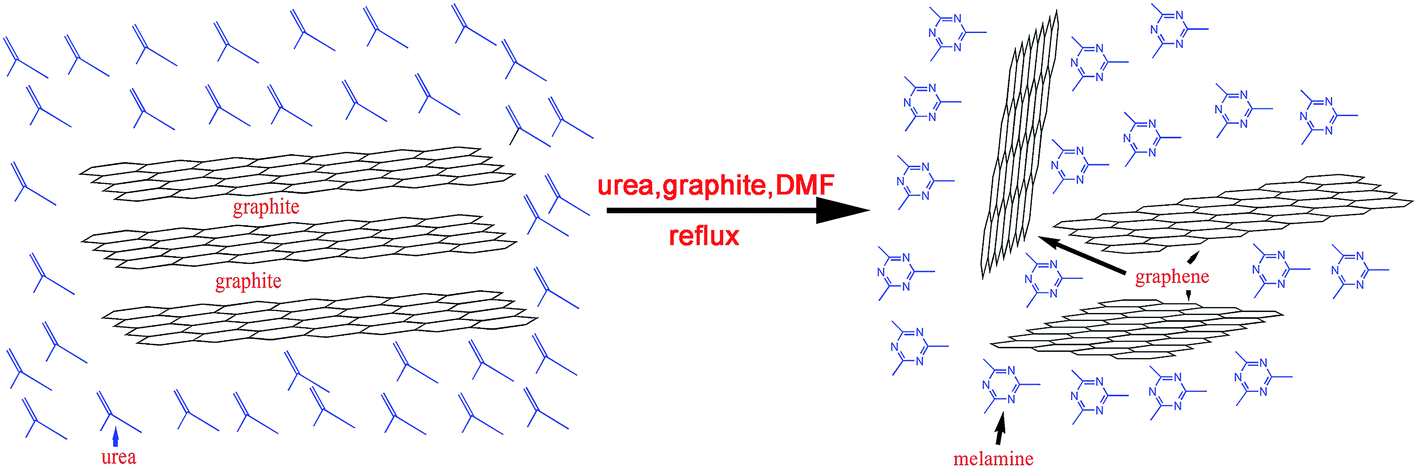 | ||
| Fig. 7 Schematic illustration for the exfoliation of graphite through a sonication-free LPE method. | ||
Sonication-free LPE assisted with urea was firstly used to exfoliate graphite into individual atomic planes. Thus, a new method for the preparation of high quality single-layer or few-layer graphene sheets with large sizes from exfoliation of graphite was achieved. This top-down approach can be easily scaled up since the yield is only dependent on the size of the vessel.
The existence of melamine in the exfoliation process of graphite was confirmed by thermogravimetric analysis. The TG and DSC curves of the graphene sample were determined directly by filling graphene powder into a thermogravimetric analysis sample pan. The graphene powder was obtained by drying the dispersion after reflux in vacuum oven at 180 °C for 5 h. Subsequently, it was heated to 800 °C at 10 °C min−1 in N2 atmosphere on a thermogravimetric analyzer to determine the mass loss of the sample. Urea unreacted may have been removed completely from the sample during the drying process, so there is not a weight loss at below 300 °C as shown in Fig. 8. However, an obvious weight loss appears at around 300 °C, which corresponds to the amount of melamine formed in reflux procedure. The graphene samples obtained by ball milling treatment with melamine presented a weight loss at around 300 °C.43 Further slight loss took place above this temperature, showing that little oxidative defects were created around the graphene sheets.
 | ||
| Fig. 8 Thermogravimetric analysis of graphene powder obtained by drying the dispersion after reflux in vacuum at 180 °C for 5 h. | ||
XPS results also support our exfoliation mechanism because the C 1s peak is similar with ones of graphene sheets produced by ball milling treatment with melamine43 but is different with those of graphene sheets prepared by sonication LPE. The wide scan and C 1s high resolution XPS were obtained for graphene sample and shown in Fig. 9. Fig. 9a shows the characteristic C 1s, N 1s, and O 1s core-level photoemission peaks at ∼285, ∼399, and ∼532 eV, respectively. Fig. 9b shows that the C 1s peak can be deconvoluted into five different components. The most intense peak at 284.60 eV is assigned to sp2 C-atoms of the graphene sheets and the component at 291.69 eV that corresponds to its p–p transitions. The two peaks are a signature of graphitic carbon. The component at 286.05 eV has been attributed to C–N bonds and the component at 289.59 eV is result from to COO species. It is worth noting that the component at 287.7 eV is due to the presence of melamine.43 This fact confirms that melamine was indeed formed during reflux of mixture of graphite powder and urea in DMF.
 | ||
| Fig. 9 (a) Wide scan XPS and (b) high resolution XPS of C 1s for graphene sheets prepared by sonication-free LPE method. | ||
3.3 Yield of graphene sheets
In order to measure graphene concentrations (CG), the as-prepared graphene dispersion in DMF was filtered onto a 20 nm alumina membrane and thoroughly washed with hot water. It is expected that unreacted urea and formed melamine were removed from dispersion because urea and melamine can be dissolved in hot water. Then graphene sheets were redispersed into tetrahydrofuran (THF) solvent. The absorbance of the dispersions should thus result completely from the dispersed graphene sheets since urea and melamine had been removed by washing. Fig. 10a shows UV-vis absorbance at 660 nm as functions of CG in THF and the inset presents absorption spectrum for a graphene dispersion at CG = 0.006 mg mL−1. The UV-vis extinction coefficient of graphene sheets in THF was determined by measuring the absorbance at 660 nm for eight reference dispersions prepared at different dilutions from a graphene dispersion whose CG was precisely determined (0.041 mg mL−1 in THF) by a thermogravimetric analysis following previous report.44 As shown in Fig. 10a, a linear dependence of the absorbance on concentration is found, achieving an extinction coefficient of 40.6 mL mg−1 cm−1 and this value is very close to the value of 42.3 mL mg−1 cm−1 (at 660 nm) determined by Ye et al.44 Thus the CG value of the dispersions can be quantified from their UV-vis absorbance at 660 nm by following the Lambert–Beer Law. | ||
| Fig. 10 (a) UV-vis absorbance at 660 nm (A660) as functions of graphene concentration (CG) for graphene sheets exfoliated by a sonication-free LPE method. Inset: representative absorption spectrum for a graphene dispersion at CG = 0.006 mg mL−1; (b) CG as a function of mass ratio of urea to graphite for graphene sheets exfoliated; (c) CG as a function of reflux time for graphene sheets exfoliated. | ||
The effects of mass ratio of urea to graphite and reflux time on CG were investigated. Fig. 10b shows the dependence of CG on the mass ratio of urea/graphite, which varies from 0 to 15 when reflux time was fixed for 12 h. An increase in CG is noted with the increase of the mass ratio from 0 to 9, which should be attributed to the increased amount of intercalating agent melamine by the conversion of urea. Further increase of the mass ratio from 10 to 15, however, shows only slight changes in CG. When mass ratio is 9 CG value is 0.15 mg mL−1. Subsequently, the effects of reflux time on CG were investigated with a fixed mass ratio of urea to graphite of 9 and the reflux time was varied from 3 h to 36 h, resulting in CG range from 0.03 to 0.20 mg mL−1 (Fig. 10c). It was found that with increasing the reflux time from 3 to 24 h CG increased while the reflux time increased to over 24 h CG had not obvious enhancement. The higher CG was obtained for 24 h, achieving to 0.19 mg mL−1 that generated by our sonication-free LPE is also compared to 0.25 mg mL−1 obtained by LPE in NMP as the best known solvent for a sonication time of 48 h.36 However, when reflux time was over 24 h the generation of many holes on graphene sheets were found. The optimal reflux time was proved to 24 h for which both CG and the integrity of the graphene sheets are available.
4. Conclusions
In summary, we introduce a novel sonication-free LPE method for fabrication of high-quality and large-size graphene sheets with single or few-layer. The successful exfoliation has been confirmed by the characterizations of the graphene sheets with TEM, AFM, Raman, and XPS techniques. These high quality graphene sheets can form flexible films and has a comparable conductivity. The urea plays an important role in the exfoliation process because it can convert into melamine that has intercalation effect, resulting in the exfoliation of graphite to generate graphene sheets. The mass ratio of urea to graphite and reflux time have important influences on graphene concentration. The overall results suggest that the sonication-free LPE method to graphene production is a scalable and low-cost route that may open a way closer to practical applications.Acknowledgements
This work was supported by Key Project of Tianjin Sci-Tech Sup-port Program (no. 08ZCKFSH01400).References
- W. Song, K. W. Kim, S. J. Chang, T. J. Park, S. H. Kim, M. W. Jung, G. Lee, S. Myung, J. Lim, S. S. Lee and K. S. An, J. Mater. Chem. C, 2015, 3, 725–728 RSC.
- A. A. Balandin, Nat. Mater., 2011, 10, 569–581 CrossRef CAS PubMed.
- A. Flores, H. J. Salavagione, F. Ania, G. Martínez, G. Ellis and M. A. Gómez-Fatou, J. Mater. Chem., 2015, 3, 1177–1180 RSC.
- T. V. Khai, H. G. Na, D. S. Kwak, Y. J. Kwon, H. Ham, K. B. Shim and H. W. Kim, Carbon, 2012, 50, 3799–3806 CrossRef PubMed.
- S. Frank, Nat. Nanotechnol., 2010, 5, 487–496 CrossRef PubMed.
- Q. Ma, X. Zhu, D. Zhang and S. Liu, J. Mater. Chem. C, 2014, 2, 8956–8961 RSC.
- K. J. Jiao, X. Wang, Y. Wang and Y. Chen, J. Mater. Chem. C, 2014, 2, 7715–7721 RSC.
- D. A. C. Brownson, D. K. Kampouris and C. E. A. Banks, J. Power Sources, 2011, 196, 4873–4885 CrossRef CAS PubMed.
- H. Kim, A. A. Abdala and C. W. Macosko, Macromolecules, 2010, 43, 6515–6530 CrossRef CAS.
- C. Huang, C. Li and G. Shi, Energy Environ. Sci., 2012, 5, 8848–8868 CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Y. Jia, Y. Wu, S. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 217–224 CrossRef CAS PubMed.
- Z. Wang, P. Li, Y. Chen, J. Liu, H. Tian, J. Zhou, W. Zhang and Y. Li, J. Mater. Chem. C, 2014, 2, 7396–7401 RSC.
- L. Chen, Y. Hernandez, X. L. Feng and K. Mllen, Angew. Chem., Int. Ed., 2012, 51, 7640–7654 CrossRef CAS PubMed.
- P. Blake, P. D. Brimicombe, R. R. Nair, T. J. Booth, D. Jiang, F. Schedin, L. A. Ponomarenko, S. V. Morozov, H. F. Gleeson, E. W. Hill, A. K. Geim and K. S. Novoselov, Nano Lett., 2008, 8, 1704–1708 CrossRef PubMed.
- J. N. Coleman, Acc. Chem. Res., 2013, 46, 14–22 CrossRef CAS PubMed.
- T. Hasan, F. Torrisi, Z. Sun, D. Popa, V. Nicolosi, G. Privitera, F. Bonaccorso and A. C. Ferrari, Phys. Status Solidi B, 2010, 247, 2953–2957 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- C. E. Hamilton, J. R. Lomeda, Z. Sun, J. M. Tour and A. R. Barron, Nano Lett., 2009, 9, 3460–3462 CrossRef CAS PubMed.
- L. Xu, J. W. McGraw, F. Gao, M. Grundy, Z. Ye, Z. Gu and J. L. Shepherd, J. Phys. Chem. C, 2013, 117, 10730–10742 CAS.
- M. D. Stoller, S. Park, Y. W. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed.
- K. Parvez, Z. S. Wu, R. J. Li, X. J. Liu, R. Graf, X. L. Feng and K. Müllen, J. Am. Chem. Soc., 2014, 136, 6083–6091 CrossRef CAS PubMed.
- L. Gong, I. A. Kinloch, R. J. Young, I. Riaz, R. Jalil and K. S. Novoselov, Adv. Mater., 2010, 22, 2694–2697 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, H. Porwal, P. May, K. Nawaz and J. N. Coleman, Carbon, 2012, 50, 470–475 CrossRef CAS PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312–1314 CrossRef CAS PubMed.
- K. S. Kim, Y. Zhao, H. Jang, S. Y. Lee, J. M. Kim, K. S. Kim, J. H. Ahn, P. Kim, J. Y. Choi and B. H. Hong, Nature, 2009, 457, 706–710 CrossRef CAS PubMed.
- A. V. Alaferdov, A. Gholamipour-Shirazi, M. A. Canesqui, Y. A. Danilov and S. A. Moshkalev, Carbon, 2014, 69, 525–535 CrossRef CAS PubMed.
- J. Geng, B. S. Kong, S. B. Yang and H. T. Jung, Chem. Commun., 2010, 46, 5091–5093 RSC.
- A. Ciesielski and P. Samorı, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- P. Gayathri, M. Jayabal, M. Kottaisamy and V. Ramakrishnanl, AIP Adv., 2014, 4, 027116 CrossRef PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- U. Khan, H. Porwal, A. O'Neill, K. Nawaz, P. May and J. N. Coleman, Langmuir, 2011, 27, 9077–9092 CrossRef CAS PubMed.
- A. A. Green and M. C. Hersam, Nano Lett., 2009, 12, 4031–4036 CrossRef PubMed.
- M. Lotya, P. J. King, U. Khan, S. De and J. N. Coleman, ACS Nano, 2010, 4, 3155–3162 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Y. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gun'Ko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563–568 CrossRef CAS PubMed.
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864–871 CrossRef CAS PubMed.
- H. Chen, M. B. Müller, K. J. Gilmore, G. G. Wallace and D. Li, Adv. Mater., 2008, 20, 3557–3561 CrossRef CAS PubMed.
- H. A. Becerril, J. Mao, Z. Liu, R. M. Stoltenberg, Z. Bao and Y. E. Chen, ACS Nano, 2008, 2, 463–470 CrossRef CAS PubMed.
- D. Li, M. B. Müller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- H. L. Guo, P. Su, X. F. Kang and S. K. Ning, J. Mater. Chem. A, 2013, 1, 2248–2255 CAS.
- J. D. Wuest and A. Rochefort, Chem. Commun., 2010, 46, 2923–2925 RSC.
- V. Leon, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. de la Hoz, M. Prato and E. Vazquez, Chem. Commun., 2011, 47, 10936–10938 RSC.
- L. V. Leo'n, M. Quintana, M. A. Herrero, J. L. G. Fierro, A. D. L. Hoz, M. Pratob and E. Va'zquez, Chem. Commun., 2011, 47, 10936–10938 RSC.
- L. Xu, J. W. McGraw, F. Gao, M. Grundy, Z. Ye, Z. Gu and J. L. Shepherd, J. Phys. Chem. C, 2013, 117, 10730–10742 CAS.
| This journal is © The Royal Society of Chemistry 2015 |