
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtraction of lithium-ion battery electrolytes with liquid and supercritical carbon dioxide and additional solvents
Martin
Grützke
,
Xaver
Mönnighoff
,
Fabian
Horsthemke
,
Vadim
Kraft
,
Martin
Winter
and
Sascha
Nowak
*
University of Münster, MEET Battery Research Center, Institute of Physical Chemistry, Corrensstraße 46, 48149 Münster, Germany. E-mail: sascha.nowak@uni-muenster.de; Fax: +49-251-83 36032
First published on 8th May 2015
Abstract
A flow-through method for the extraction of lithium-ion battery electrolytes with supercritical and liquid carbon dioxide (sc and liq CO2) under the addition of different solvents has been developed and optimized to achieve quantitative extraction of the electrolyte from commercial LiNi1/3Co1/3Mn1/3O2 (NMC)/graphite 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 650 cells. Furthermore, the time-dependence of the extraction procedure was investigated and demonstrated. The extracts were analyzed with gas and ion chromatography. Linear carbonates like dimethyl carbonate (DMC) and ethyl methyl carbonate (EMC), respectively, were better extracted with liq CO2, whereas the cyclic carbonate ethylene carbonate (EC) was recovered in higher amounts with sc CO2. The addition of solvents to the CO2 resulted in improved recovery for all the ingredients but most effectively for LiPF6, which could not be obtained by extraction with CO2 only. The best results were achieved by extracting for 30 minutes with liq CO2 (25 °C, 60 bar) and 0.5 mL min−1 acetonitrile (ACN)/propylene carbonate (PC) in a mixture of three to one and an additional 20 minutes with liq CO2 only, to yield (89.1 ± 3.4) wt% electrolyte in almost its original composition of DMC, EMC, EC (1:1:1) with 1.1 mol L−1 LiPF6. Therefore, the presented method can be relevant for the recycling of lithium ion battery electrolytes but has to be validated for up-scaled processes. Furthermore, the suitability of CO2 extraction as a tool for post-mortem or aging investigations of LIB electrolytes could once more successfully be demonstrated due to the extraction of aging products like diethyl-2,5-dioxahexane dicarboxylate (DEDOHC) from a pouch cell, which was electrochemically aged for 1000 cycles at 1 C. In this context, extraction times and recovery rates were drastically improved compared to our previously reported static extraction experiments.
650 cells. Furthermore, the time-dependence of the extraction procedure was investigated and demonstrated. The extracts were analyzed with gas and ion chromatography. Linear carbonates like dimethyl carbonate (DMC) and ethyl methyl carbonate (EMC), respectively, were better extracted with liq CO2, whereas the cyclic carbonate ethylene carbonate (EC) was recovered in higher amounts with sc CO2. The addition of solvents to the CO2 resulted in improved recovery for all the ingredients but most effectively for LiPF6, which could not be obtained by extraction with CO2 only. The best results were achieved by extracting for 30 minutes with liq CO2 (25 °C, 60 bar) and 0.5 mL min−1 acetonitrile (ACN)/propylene carbonate (PC) in a mixture of three to one and an additional 20 minutes with liq CO2 only, to yield (89.1 ± 3.4) wt% electrolyte in almost its original composition of DMC, EMC, EC (1:1:1) with 1.1 mol L−1 LiPF6. Therefore, the presented method can be relevant for the recycling of lithium ion battery electrolytes but has to be validated for up-scaled processes. Furthermore, the suitability of CO2 extraction as a tool for post-mortem or aging investigations of LIB electrolytes could once more successfully be demonstrated due to the extraction of aging products like diethyl-2,5-dioxahexane dicarboxylate (DEDOHC) from a pouch cell, which was electrochemically aged for 1000 cycles at 1 C. In this context, extraction times and recovery rates were drastically improved compared to our previously reported static extraction experiments.
1. Introduction
Besides its well-known role as a greenhouse gas in climate change discussions,1–4 carbon dioxide also takes part in many chemical reactions. It is for example used in the synthesis of linear5–8 and cyclic9 organic carbonates, which are, inter alia, solvents for conducting salts, typically LiPF6,10 in modern lithium-ion batteries (LIBs).11,12 CO2 has also been used as a solid electrolyte interphase (SEI)13 forming electrolyte additive in lithium metal and lithium-ion batteries14 and is produced when the organic carbonate based electrolyte solvents are oxidized, e.g. during overcharge15 in some cases. CO2 is also generated during the reduction of organic carbonate based battery electrolytes.16,17 Furthermore, graphite anode surface modification with CO2 has been performed.18,19 Moreover, CO2 is widely applied as a monomer in materials synthesis20 or a C-1 block in the synthesis of polycarbonates21 and other organic products.22In addition to its gaseous, liquid and solid phase, carbon dioxide shows a relatively easy available supercritical phase after adjusting temperature and pressure above the critical point which is at 31 °C and 74 bar.23 The dissolving properties of sc CO2 are comparably high for many organic substances, especially in combination with additional solvents. Thus, CO2 is the most applied extraction medium in supercritical fluid extraction (SFE).24 The most prominent example for the application of sc CO2 extraction might be the decaffeination process of coffee.25,26 Likewise, sc CO2 is generally used a lot as extraction medium in food chemistry.27–29 Furthermore, its usage as eluent is also important in supercritical fluid chromatography (SFC),24 as drying agent in crack free silica aerogel production30,31 and as reaction medium in olefin polymerization reactions32 or metal nanoparticle synthesis.33
Sloop et al. described the extraction of electrolytes from energy storage devices with supercritical fluids.34 LIBs were mentioned in this patent but detailed information about the extraction behavior were rare. LIBs are the most applied energy storage systems for modern consumer and portable electronics like smartphones and cameras, and the most promising battery technology for electric or hybrid electric vehicles.35,36 LIBs usually consist of a graphitic anode and a transition metal oxide77 or phosphate cathode78 with a polymeric or ceramic separator in between. A non-aqueous aprotic electrolyte serves for charge transfer in form of lithium-ions. Electrolytes for LIBs are made of different linear carbonates (e.g. dimethyl carbonate (DMC) and ethyl methyl carbonate (EMC)) in a mixture with ethylene carbonate (EC).11,12,37 LiPF6 is generally used as conducting salt and dissolved in this solution in concentrations of about 1 mol L−1. Moreover, additives for various electrolyte properties can be present.38,39 Dimethyl methylphosphonate can be applied as flame retardant,40 cyclohexylbenzene for overcharge protection11 and vinylene carbonate as film forming additive for controlled formation of the SEI.11,12,41–46
Different aging processes occur in LIBs which lead to a creeping loss of capacity and a limited cell lifetime.47 One of these aging effects is the decomposition of the electrolyte during cycling which is furthermore influenced by the temperature and the quality of each and every compound in the cell, especially with regards to protic impurities. Research on electrolyte decomposition as well as the development of new and specific analytical methods has been done in recent years,38,48–67 but there is still a far way to go until the complete picture of electrolyte aging in LIBs is fully understood. Furthermore, the quantitatively extraction from the LIBs is of interest for recycling processes. Since 50 wt% of a cell has to be recovered, the electrolyte represents a target component for the recycling of LIBs. Additionally, fluoride or fluorinated compounds can hamper or damaged industrial scaled recycling processes and have to be removed as well.
We recently reported the extraction of LIB electrolytes with supercritical helium head pressure carbon dioxide (sc HHPCO2) in a simple autoclave setup.63 A successful proof of principle experiment was demonstrated. Furthermore, the sc HHPCO2 extraction could be presented as sample preparation tool for LIB post-mortem aging investigations. HHPCO2 was used in these experiments to keep the experimental equipment as easy as possible. Sc CO2 conditions can be achieved without a compressor already from the bottle. Nevertheless, HHPCO2 is more expensive than conventional liq CO2 with the same purity. Furthermore, the recovery rate for the electrolyte from the investigated 18650 cells was relatively low (<10 wt%). Moreover, it was not possible to obtain the conducting salt but only the organic carbonates. To overcome these drawbacks and to further investigate the extraction of LIB electrolytes with CO2 we installed a conventional flow-through SFE extraction unit. Herein, we report the results for the extraction of commercial 18650 cells (2.2 Ah) as well as in-house-made pouch cells (4.8 Ah), both based on LiNi1/3Co1/3Mn1/3O2 (NMC)/graphite electrode chemistry, with liquid and supercritical carbon dioxide regarding the recovery rate of the particular electrolyte components, additional solvents in the CO2-stream and the time dependency of the extraction procedure. Since, one investigation was the recycling and aging of LIBs, we decided to use electrolytes which are state-of-the-art in commercial applied LIBs. Nevertheless, it is still possible to investigate alternative systems.
2. Experimental
2.1. Chemicals and materials
Panasonic CGR18650CH Li-ion MH12210 (2.2 Ah, NMC/graphite) were bought from BattEnergy. 18650 cells are often used for safety investigations,68–70 and are generally applied in laptops and E-bikes. 4.8 Ah NMC/graphite pouch cells were fabricated with an in-house battery line. Anhydrous acetronitrile (ACN) (99.8%) from Sigma-Aldrich, and dry and fresh battery grade diethyl carbonate (DEC; 99.9%) and propylene (PC) (99.9%) from BASF were used for solvent addition in the CO2 extraction experiments. DMC (99.9%), EMC (99.0%), DEC (99.9%) and EC (99.9%) for gas chromatography (GC) experiments, tartaric acid (99.5%), dipicolinic acid (99.0%), Na2CO3 (Certipur), NaHCO3 (99.7%) and H2SO4 (0.1 mol L−1) were purchased from Merck. Aqueous lithium standard solution (TraceCert IC standard, 1 g L−1 Li+, ≤0.1% HNO3) and aqueous fluoride standard solution (TraceCert IC standard, 1 g L−1 NaF) were supplied by Fluka. ACN (99.9%, HiPerSolv CHROMANORM, LC-MS grade) from VWR and Milli Q water, produced with an in-house Millipore filter system were used for sample dilution. CO2 (purity 5.0) for SFE and helium (purity 6.0) for GC experiments were purchased from Westfalen Gas.
2.2. Cell cycling procedures
The pouch cells were cycled with a Maccor Series 4000 Battery Tester 20A in an environmental test chamber (Binder KB 400) at 20 °C. The cells were charged with 0.2 C in constant current mode up to a voltage of 4.2 V, followed by a constant voltage step at 4.2 V for 30 min. The discharge was performed with 1 C in constant current mode down to a voltage of 3.0 V. Subsequently, the cell was charged and discharged with 1 C (constant current) between 3.0 V and 4.2 V for 1000 cycles. Before opening the cells an extended discharge step was carried out, composed of successive and decreasing constant current steps each until a voltage of 3.0 V and constant rest periods between the discharge steps.2.3. Sample preparation
The discharged cells (18650 and pouch) were frozen at −18 °C for at least 10 h, so that electrolyte evaporation during cell opening is reduced to a minimum, and subsequently opened in a glove box (O2, H2O < 0.1 ppm) by slicing the two ends of the metal shell with an industrial cutter (18650 cells), similar to the self-made cutter of Aurbach et al.,71 or by cutting the outer pouch foil with ceramic scissors (pouch cells). The jelly role as well as the stack, were removed of the shell, uncoiled and put, as dense as possible, into the pressure chamber of the extraction equipment.
2.4. Extraction procedure
After connecting the extraction chamber to the CO2 supply and adjusting pressure and temperature in the chamber to extraction conditions (liquid: 25 °C, 60 bar; supercritical: 40 °C, 300 bar), the experiment begun with a static (non-flow-through) equilibration step which was hold for 30 min, followed by 2 min of CO2 flow-through with subsequent 5 min static (non-flow-through) equilibration, in which the amount of collected extract was weighted (experiments in Fig. 1 and 2). The outlet valve of the CO2-extraction-stream was heated to 55 °C and the electrolyte was collected with a cryogenic trap of aluminum (−78 °C) behind the outlet valve. | ||
| Fig. 1 Time dependency of the amount of recovered electrolyte from commercial 18650 cells after formation extracted with supercritical (300 bar, 40 °C; red stars) and liquid (60 bar, 25 °C; black squares) CO2. | ||
 | ||
| Fig. 2 Compositions of the recovered electrolyte from commercial 18650 cells after formation extracted with supercritical (A) and liquid (B) CO2 for the first three data points of Fig. 1 determined with GC-MS. Blue, top: EC; yellow, middle: EMC; magenta, bottom: DMC. | ||
For the results in Fig. 3–6, additional solvent (ACN, DEC, PC) with a flow of 0.5 mL min−1 was added to the CO2-stream. After an equilibration step of 30 min at the beginning with CO2, but without solvent, it was extracted in flow-through for two minutes with subsequent 5 min of static equilibration time. The (2 + 5) min step was repeated 15 times, followed by 20 min flow-through extraction without solvent (only CO2).
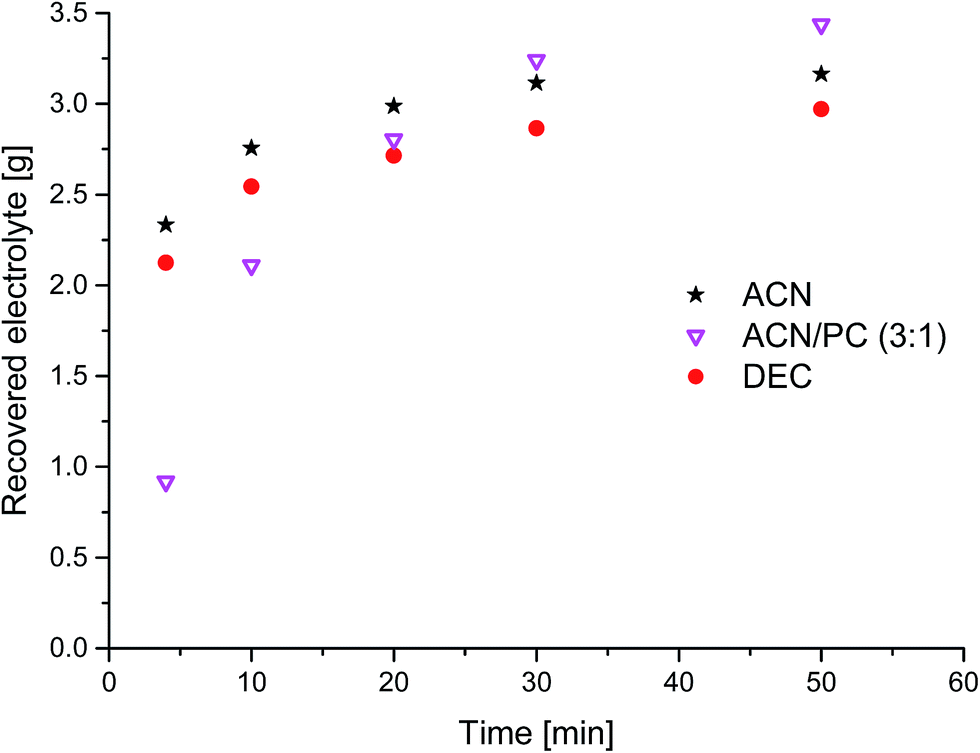 | ||
| Fig. 3 Time dependency of the amount of recovered electrolyte from commercial 18650 cells after formation extracted with liquid CO2 and 0.5 mL min−1 additional solvents (black stars: ACN; magenta triangles: ACN–PC (3:1); red circles: DEC). | ||
2.5. Analytical equipment
650 cells and 100 mL for pouch cells) and all gas lines consist of stainless steel. At the beginning of each extraction experiment, the device was investigated for gas leaks. Furthermore, the CO2 flow was adjusted to (15 ± 3) L min−1.
Samples were diluted 1:10 to 1:50 with water for anion analysis and 20 μL were injected. An aqueous eluent with 1.8 mmol L−1 Na2CO3 and 1.7 mmol L−1 NaHCO3 was used with a flow of 1 mL min−1 on a Metrosep A Supp 4-250/4.0 + Metrosep A Supp 4/5 Guard/4.0 column at a temperature of 40 °C. After 10 min, a gradient step was applied and over 3 min, ACN was added up to a concentration of 50 vol%. F− could be confirmed via the retention time of a standard solution. PO2F2−, PF6− and alkyl fluoro phosphates were identified with IC hyphenated to electrospray ionization-mass spectrometry (ESI-MS). Equipment, procedure and conditions were recently published and applied.60,64,65
:100, ACN) was injected at an injection temperature of 230 °C. The system was run with helium as carrier gas with a column flow of 1 mL min−1, a purge flow of 3 mL min−1, a split of 1:100 and the following column oven program: starting with 40 °C for 2 min, the temperature was increased with a rate of 30 °C min−1 to 230 °C and held for 2.67 min. It was measured in the electron ionization (EI) mode at an ion source temperature of 200 °C, a filament voltage of 70 V, a detector voltage, which was chosen relative to the particular tuning result (∼1 kV) in a range of 25–300 m/z with an event time of 0.1 s and a GC-MS interface temperature of 250 °C.
Quantitative results were obtained with appropriate dilution of the samples to be within the external five-point calibration (R2 > 0.999) in a range of 10 mg kg−1 to 1500 mg kg−1 (10 mg kg−1 to 100 mg kg−1 for DEDOHC) under the same GC-MS conditions as just described but with single ion monitoring (SIM) via the following ions: DMC (59 m/z), EMC (77 m/z), DEC (91 m/z), PC (102 m/z), EC (88 m/z) and DEDOHC (89 m/z). Each sample and concentration was measured two times. Limits of detection (LOD) and limits of quantification (LOQ) were determined according to DIN 32645: DMC (31.7 mg L−1, 121.0 mg kg−1), EMC (14.9 mg kg−1, 56.9 mg kg−1), DEC (12.0 mg kg−1, 46.3 mg kg−1), PC (11.7 mg kg−1, 49.4 mg kg−1), EC (13.3 mg kg−1, 51.1 mg kg−1) and DEDOHC (3.6 mg kg−1, 14.0 mg kg−1).
3. Results and discussion
First of all, it was necessary to develop a suitable extraction method to extract and collect a LIB electrolyte with the Applied Separations Spe-ed SFE System extraction equipment. We started by putting a LIB electrolyte (each experiment the same amount) containing polyethylene fleece separator into the extraction chamber of the extraction equipment. For the development of an appropriate collection method, the sample was always extracted under the same supercritical conditions: 40 °C, 80 bar CO2 in flow-through mode for one hour. The outlet valve of the extraction device is heated, which is necessary due to the Joule–Thomson-Effect.73 This temperature was set to 55 °C because of the possible thermal decomposition of LiPF6 at higher temperature.74 It was proven, that collecting the electrolyte from the CO2-stream during pressure relaxation with a cryogenic trap (aluminum vial at −78 °C) resulted in better recovery of the electrolyte than washing solutions with different organic solvents (e.g. ACN). Furthermore, it is advisable to pack the sample in the extraction chamber as dense as possible to get good extraction results, otherwise the CO2, as extraction medium, just passes by the sample without leaching it.After adjusting these first parameters, further method development was done with 2.2 Ah, NMC/graphite commercial 18650 cells (Panasonic CGR18650CH Li-ion MH12210). The uncoiled jelly roll was densely packed into the extraction chamber. Variation of the pressure at supercritical extraction conditions (40 °C, 80 bar vs. 300 bar) resulted only in slightly different recovery rates at the same CO2 flow, whereas variation of the flow at the same pressure gave much higher recovery rates (by weight) in the same time. Unfortunately this parameter was limited due to the mentioned Joule–Thomson-Effect because CO2 simply freezes out in the cryogenic trap when the flow is too high. Thus, the flow was adjusted to 15 L min−1 at the outlet valve for all further experiments as good compromise between high flow rate (which means short extraction times) and resublimating CO2. The CO2 flow control of the outlet valve over time showed, that the used extraction equipment could not better be adjusted than within 20% relative standard deviation which means a CO2 flow of (15 ± 3) L min−1.
Two different CO2 conditions were compared. While extracting with liq CO2 (25 °C, 60 bar), we reproducibly achieved a higher amount of extracted electrolyte at the same extraction time and volumetric flow as with sc CO2 (40 °C, 300 bar). This may be explained by differences due to the flow behavior of liquid CO2 compared to sc CO2 (laminar vs. non-laminar). We further investigated this behavior in a time-resolved experiment (Fig. 1), which confirmed our results. Most of the extracted amount is obtained in the first minutes. Also the better recovery with liq CO2 already happens at the beginning of the extraction. The demonstrated data show exponential behavior over time. and therefore the deviation during. Analyses of the extracts of the first minutes with GC-MS reveal that the linear carbonates, DMC and EMC, are extracted with higher efficiency than EC (Fig. 2). Interestingly, the higher amount which is achieved with liq CO2 is only linear carbonates. Compared to liq CO2, extraction with sc CO2 is better suited for EC. CO2 is a relatively nonpolar extraction medium26 which explains why the more polar EC (dipole moment: 4.61 D) is less well extracted than DMC (0.76 D) and EMC (0.89 D).11 This nonpolarity is also the reason for the poor extraction of the conducting salt which could only be gained in trace amounts (as confirmed by IC), which was also observed for the extraction of the same 18650 cells with sc HHPCO2 with a different experimental setup.63 The amount of electrolyte in the investigated 18650 cells was determined to 3.76 g by drying an opened cell at 120 °C in vacuum over 10 hours. We concluded that this amount could not be reached under the so far applied extraction conditions. Only the linear carbonates could quantitatively be recovered after a very long extraction time (over 3 h).
To overcome (i) these drawbacks of long extraction times, (ii) the small concentration of EC and (iii) the not extracted conducting salt, extraction experiments with added solvents to the liq CO2 were realized. The time-resolved results for three different additional solvents are summarized in Fig. 3. The exponential behavior ends at 50 min of extraction time at recovery rates which are much higher compared to extraction with sc CO2 or liq CO2 without additional solvents. The experiments with ACN and DEC show a similar trend with high recovery rates in the first minutes compared to the ACN/PC mixture, which leads to an indolent extraction at first, but finishes with the best overall result. An experiment with only PC as added solvent was also carried out but the collected extract was with about 2 g after the applied extraction method of 50 min much less than the three scenarios demonstrated in Fig. 3. Furthermore, a large amount of the PC apparently remained in the solid battery material during this time, which could be confirmed by weight and also by the 'wet' solid battery material after the extraction chamber was opened for cleaning purposes subsequent to the extraction experiment. Thus, it can be concluded that the addition of very polar solvents (dipole moment PC: 4.81 D)11 is only reasonable when additional solvents with medium polarity are present, that act as “moderator” between very polar solvent and the rather unpolar CO2.
Fig. 4 shows the compositions of the extracts over the whole period of extraction with DEC as additional solvent. The trend is identical to the extraction process without additional solvent. The linear carbonates (DMC and EMC) are extracted well in the first minutes, but the absolute amount is almost doubled compared to extraction with CO2 only. Furthermore, also high recoveries of EC and even of the conducting salt LiPF6 could be achieved. Fig. 5 demonstrates the GC-MS measurements corresponding to the compositions of the volatile components in Fig. 4. It can be seen that the linear carbonates are only significantly extracted in the first 20 min, whereas EC is present in the same magnitude till the end of the extraction experiment. Fig. 6 summarizes the amounts of recovered electrolyte and their compositions for the three discussed scenarios (see Fig. 3). Additionally, the composition of a reference electrolyte is shown. The reference data are the result of the quantitative investigation of electrolyte (10 μL) which was collected during cell opening. The resulted weight percentages are normalized to the amount of electrolyte per 18650 cell. In all cases of extraction with liq CO2 and additional solvents, the linear carbonates could be recovered quantitatively within the limits of error; EC and LiPF6 could be retrieved at least qualitatively. The best overall recovery rate was achieved for the added ACN/PC mixture, with (89.1 ± 3.4) wt% and furthermore with the highest concentrations for EC and LiPF6.
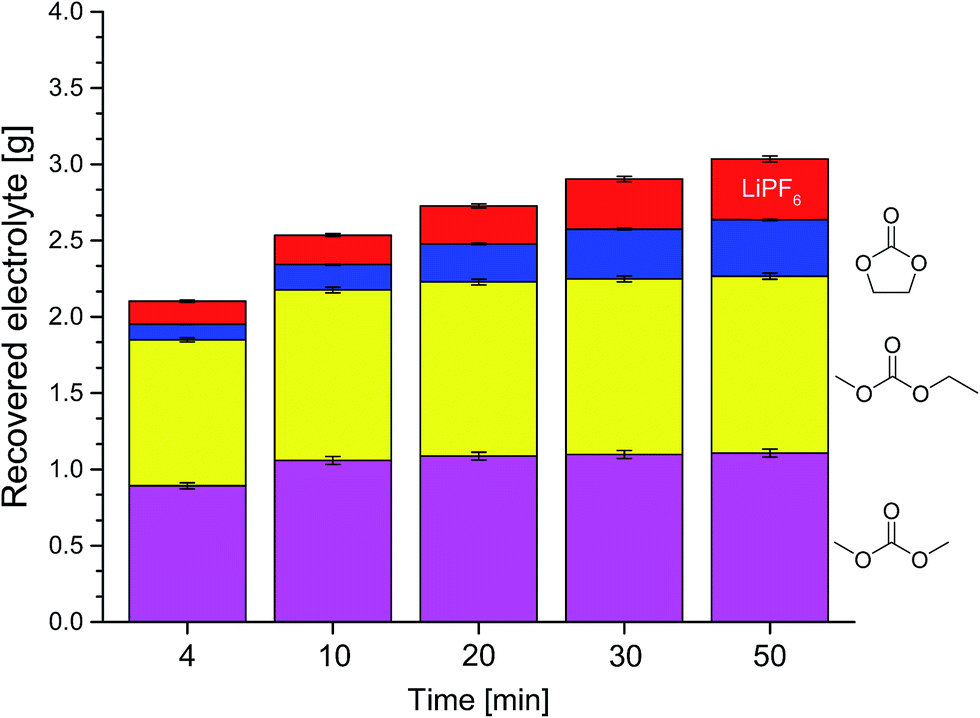 | ||
| Fig. 4 Compositions of the recovered electrolyte from commercial 18650 cells after formation extracted with liquid CO2 and additional DEC in the CO2-stream determined with GC-MS and IC corresponding to Fig. 3. Red, top: LiPF6; blue, below: EC; yellow, middle: EMC; magenta, bottom: DMC. | ||
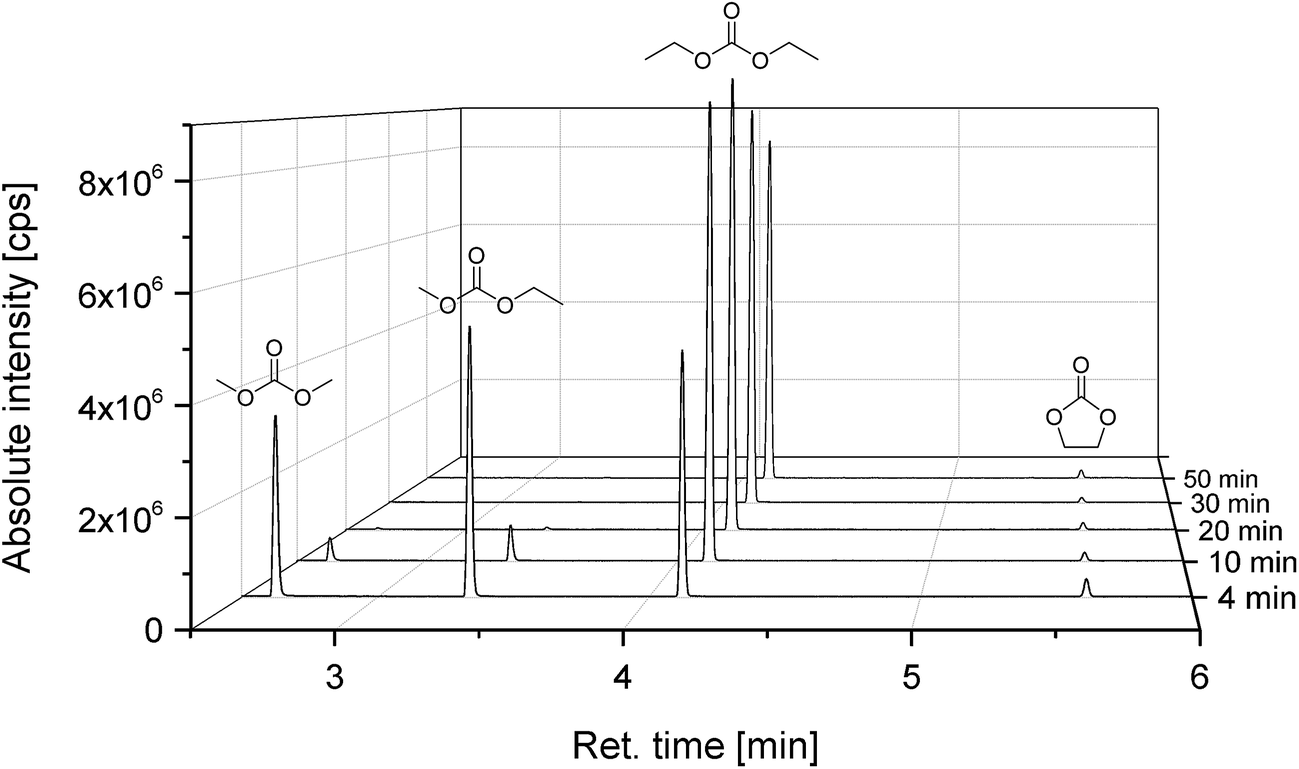 | ||
| Fig. 5 Chromatograms (GC-MS) of the time-resolved electrolyte recovery which was extracted with liquid CO2 and additional DEC, corresponding to Fig. 3 and 4. | ||
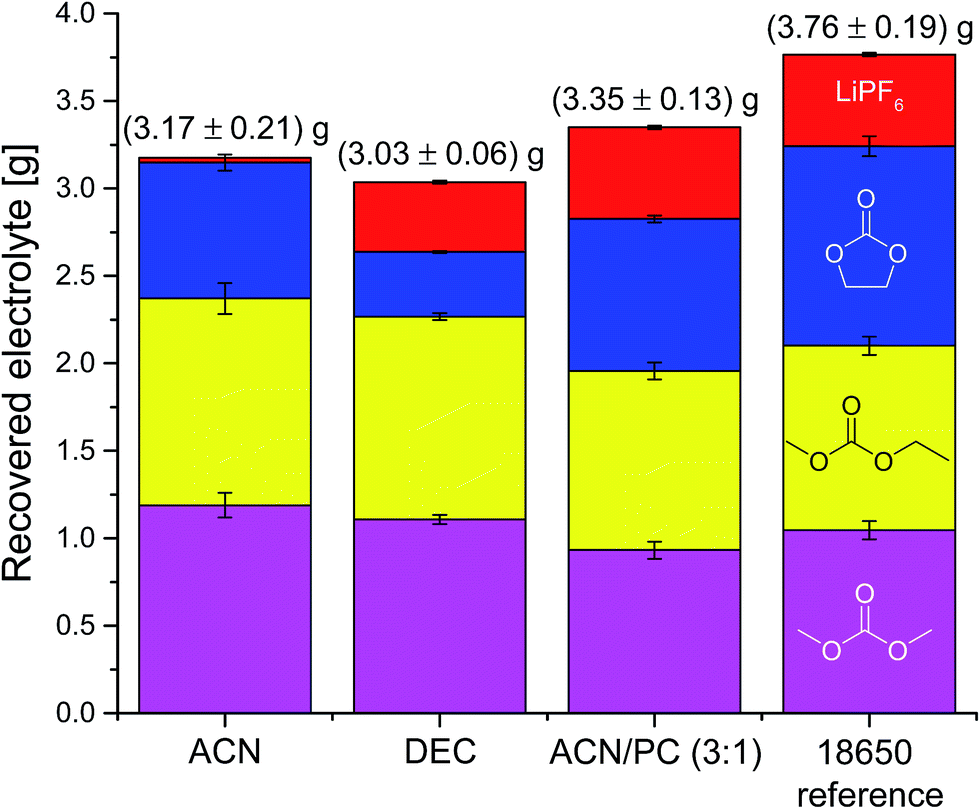 | ||
| Fig. 6 Compositions and amounts (determined with GC-MS and IC) of the recovered electrolytes from commercial 18650 cells after formation extracted with liquid CO2 and additional solvents for 30 min, with subsequent 20 min without additional solvent. Red, top: LiPF6; blue, below: EC; yellow, middle: EMC; magenta, bottom: DMC. | ||
Finally, we also investigated self-made pouch cells (graphite/NMC) after formation and after “electrochemical aging”, which were bigger than the so far used 18650 cells (4.8 Ah vs. 2.2 Ah). The results of the cycling experiment (=electrochemical aging) for 1000 charge/discharge cycles at a rate of 1 C are displayed in Fig. 7. An electrolyte consisting of 1 mol L−1 LiPF6 in a DEC, PC, EC, VC mixture was applied (quantitative composition see Table 1). All components, except VC, could be detected in the extract, which was achieved with liq CO2 and ACN as additional solvent. PC as additional solvent was not applied in this case due to its presence in the cells and therefore possible difficulties in quantification of the extract. VC had presumably been consumed during the formation cycle and thus been involved in the SEI formation.11,55 In addition to the deployed electrolyte components, DEDOHC, which is an electrolyte aging product, whose formation was already intensively discussed in literature,48,51,54–58,63,66 could clearly be identified. This confirms the result of our previous study, that CO2 can be applied as extraction agent for post-mortem or aging investigations. Fig. 8 shows the GC-MS chromatograms corresponding to the aged cell and, consequently, the extract compositions of the collected fractions. The same picture was achieved for the cell after formation. The findings for DEC, EC and PC are in good agreement to the results of the 18650 cells.
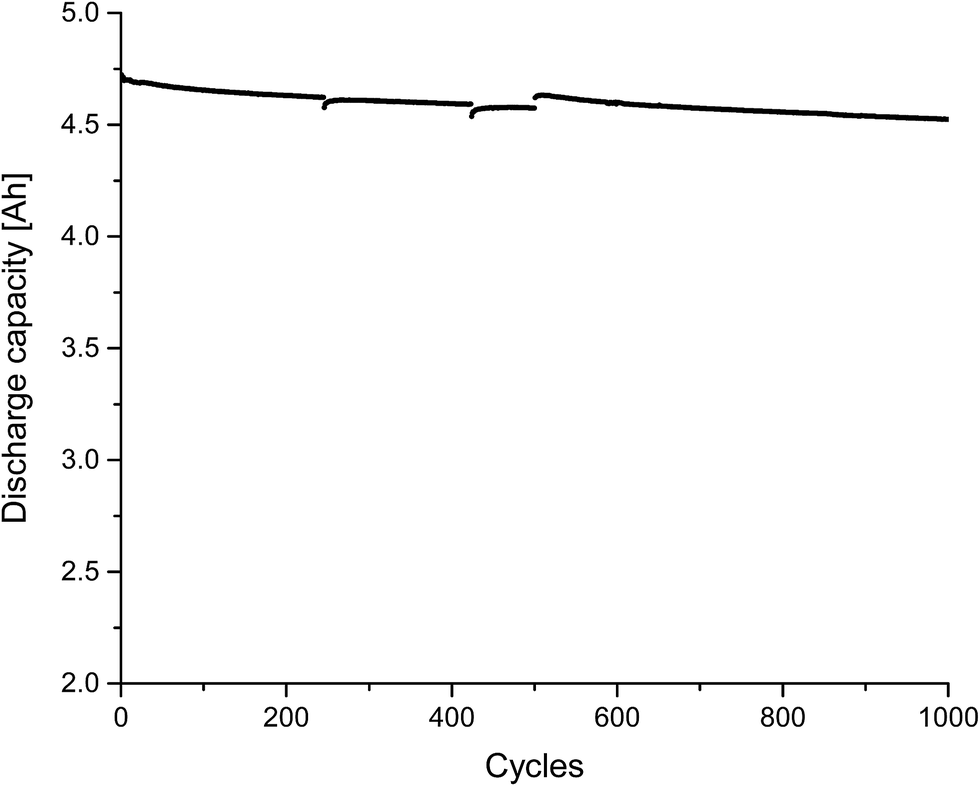 | ||
| Fig. 7 Discharge capacities for an in-house-made 4.8 Ah pouch cell (cycled at room temperature at a rate of 1 C between 3.0 V and 4.2 V). | ||
| DEC | PC | EC | VC | LiPF6 |
|---|---|---|---|---|
| 46.9 ± 2.2 | 4.5 ± 0.1 | 32.4 ± 0.7 | 2.0 ± 0.1 | 11.7 ± 0.3 |
 | ||
| Fig. 8 Chromatograms (GC-MS) of the time-resolved electrolyte recovery from a cycled (=electrochemically aged) pouch cell (Fig. 7) which was extracted with liquid CO2 and additional ACN. The absolute intensity is amplified by one order of magnitude for the last minute of retention time. | ||
DEDOHC could be extracted from the cells after formation and cycling, in the same amount of (0.4 ± 0.1) wt% within the first four minutes. Therefore, it is concluded, that DEDOHC is only formed during formation. The 18650 cell showed a different aging behavior in this case. The concentrations of DEDOHC and its derivatives were in correlation with an ongoing film growth during cycling.63 The reason for this difference could be the presence or absence, respectively of VC as electrolyte additive for controlled SEI formation.11 On the other hand, the pouch cells showed a state-of-health (SOH) of over 97% after 1000 cycles (Fig. 7), compared to 70% SOH for the 18650 cells, which makes a quantitative comparison of the two cell types difficult.
Moreover, we could detect traces of decomposition products of the conducting salt LiPF6, such as F−, PO2F2− and different fluoro phosphates with IC and IC hyphenated to electrospray ionization mass spectrometry (ESI-MS) in all samples, even for the cells, which were extracted without additional solvents. This confirms once more the suitability of CO2 extraction as sample preparation tool for post-mortem or aging investigations of LIB electrolytes.
4. Conclusions
The extraction behavior of supercritical and liquid carbon dioxide with and without additional solvents has been investigated in flow-through experiments regarding the recovery of electrolytes from lithium-ion batteries. The extracts were analyzed with GC-MS and IC for their compositions. For an electrolyte containing LiPF6, DMC, EMC and EC, extraction with liq CO2 resulted in higher recovery for linear carbonates (DMC, EMC) and lower for EC but in a higher overall yield compared to extraction with sc CO2. The extraction of the electrolyte ingredients was improved by application of additional solvents to the CO2 and, furthermore, also LiPF6 was extracted. Maximum recovery rates were achieved with mixtures of different solvents. It was demonstrated, that the electrolyte of a Panasonic 18650 cell was recovered in (89.1 ± 3.4) wt% almost quantitatively with liq CO2 and ACN–PC (3:1) as additional solvents. Thus, we conclude, that it is generally possible to apply CO2 extraction to used battery cell electrolytes by properly adjusting the extraction conditions (especially the additional solvents) to the individual cell components (electrode materials, separator, electrolyte and geometry) which should be extracted. This promising procedure could be an alternative for distortion of the electrolyte in LIB recycling,75 and even for solvent extraction.76 However, the developed method is a suitable tool for LIB electrolyte and post-mortem or aging investigations because a qualitative overview can already be achieved after a few minutes of extraction also for apparently ‘dry’ cells, where the electrolyte is deeply penetrated in the solid battery material. The application of this tool for a screening of different aged cells from several battery suppliers is currently in preparation.
Acknowledgements
We kindly acknowledge the Federal Ministry for the Environment, Nature Conservation and Nuclear Safety for funding of the project LithoRec II (project grant number: 16EM1025) in which the results in this manuscript could be achieved.References
- S. Pan, H. Tian, S. R. S. Dangal, Q. Yang, J. Yang, C. Lu, B. Tao, W. Ren and Z. Ouyang, Earth's Future, 2015, 3, 15–35 CrossRef CAS PubMed.
- Z. Morales-Reyes, J. M. Perez-Garcia, M. Moleon, F. Botella, M. Carrete, C. Lazcano, R. Moreno-Opo, A. Margalida, J. A. Donazar and J. A. Sanchez-Zapata, Sci. Rep., 2015, 5, 7811 CrossRef CAS PubMed.
- S. M. Rahman and G. A. Kirkman, Energ. Econ., 2015, 47, 129–141 CrossRef PubMed.
- J. Hansen, M. Sato, P. Kharecha, D. Beerling, R. Berner, V. Masson-Delmotte, M. Pagani, M. Raymo, D. L. Royer and J. C. Zachos, Open Atmos. Sci. J., 2008, 2, 217–231 CrossRef CAS.
- S. Fang and K. Fujimoto, Appl. Catal., A, 1996, 142, L1–L3 CrossRef CAS.
- N. S. Isaacs, B. O'Sullivan and C. Verhaelen, Tetrahedron, 1999, 55, 11949–11956 CrossRef CAS.
- T. Sakakura, J.-C. Choi, Y. Saito and T. Sako, Polyhedron, 2000, 19, 573–576 CrossRef CAS.
- S. Camy, J. S. Pic, E. Badens and J. S. Condoret, J. Supercrit. Fluids, 2003, 25, 19–32 CrossRef CAS.
- A. Buonerba, A. De Nisi, A. Grassi, S. Milione, C. Capacchione, S. Vagin and B. Rieger, Catal. Sci. Technol., 2015, 5, 118–123 CAS.
- R. W. Schmitz, P. Murmann, R. Schmitz, R. Müller, L. Krämer, J. Kasnatscheew, P. Isken, P. Niehoff, S. Nowak, G.-V. Röschenthaler, N. Ignatiev, P. Sartori, S. Passerini, M. Kunze, A. Lex-Balducci, C. Schreiner, I. Cekic-Laskovic and M. Winter, Prog. Solid State Chem., 2014, 42, 65–84 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303–4418 CrossRef CAS.
- K. Xu, Chem. Rev., 2014, 114, 11503–11618 CrossRef CAS PubMed.
- H. Schranzhofer, J. Bugajski, H. J. Santner, C. Korepp, K. C. Möller, J. O. Besenhard, M. Winter and W. Sitte, J. Power Sources, 2006, 153, 391–395 CrossRef CAS PubMed.
- J. O. Besenhard, M. W. Wagner, M. Winter, A. D. Jannakoudakis, P. D. Jannakoudakis and E. Theodoridou, J. Power Sources, 1993, 44, 413–420 CrossRef CAS.
- C. Dippel, R. Schmitz, R. Müller, T. Böttcher, M. Kunze, A. Lex-Balducci, G.-V. Röschenthaler, S. Passerini and M. Winter, J. Electrochem. Soc., 2012, 159, A1587–A1590 CrossRef CAS PubMed.
- M. Winter and P. Novák, J. Electrochem. Soc., 1998, 145, L27–L30 CrossRef CAS PubMed.
- M. Winter, R. Imhof, F. Joho and P. Novák, J. Power Sources, 1999, 81–82, 818–823 CrossRef CAS.
- H. Buqa, R. I. R. Blyth, P. Golob, B. Evers, I. Schneider, M. V. Santis Alvarez, F. Hofer, F. P. Netzer, M. G. Ramsey, M. Winter and J. O. Besenhard, Ionics, 2000, 6, 172–179 CrossRef CAS.
- H. Buqa, P. Golob, M. Winter and J. O. Besenhard, J. Power Sources, 2001, 97–98, 122–125 CrossRef CAS.
- W. Xia, S. I. Vagin and B. Rieger, Chem.–Eur. J., 2014, 20, 15499–15504 CrossRef CAS PubMed.
- P. Altenbuchner, S. Kissling and B. Rieger, in Transformation and Utilization of Carbon Dioxide, ed. B. M. Bhanage and M. Arai, Springer, Berlin Heidelberg, 2014, pp. 163–200 Search PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 3091 Search PubMed.
- D. R. Lide, CRC Handbook of Chemistry and Physics, 85 edn, 2004 Search PubMed.
- M. C. Henry and C. R. Yonker, Anal. Chem., 2006, 78, 3909–3916 CrossRef CAS PubMed.
- E. Lack and H. Seidlitz, in Extraction of Natural Products Using Near-Critical Solvents, ed. M. B. King and T. R. Bott, Springer, Netherlands, 1993, pp. 101–139 Search PubMed.
- E. J. Beckman, Environ. Sci. Technol., 2002, 36, 347A–353A CrossRef CAS.
- N. K. Sinha, D. E. Guyer, D. A. Gage and C. T. Lira, J. Agric. Food Chem., 1992, 40, 842–845 CrossRef CAS.
- J. P. Friedrich, G. R. List and A. J. Heakin, J. Am. Oil Chem. Soc., 1982, 59, 288–292 CrossRef CAS.
- E. Reverchon and F. Senatore, J. Agric. Food Chem., 1994, 42, 154–158 CrossRef CAS.
- M. J. van Bommel and A. B. de Haan, J. Non-Cryst. Solids, 1995, 186, 78–82 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4266 CrossRef CAS PubMed.
- D. Guironnet, I. Göttker-Schnetmann and S. Mecking, Macromolecules, 2009, 42, 8157–8164 CrossRef CAS.
- S. Moisan, V. Martinez, P. Weisbecker, F. Cansell, S. Mecking and C. Aymonier, J. Am. Chem. Soc., 2007, 129, 10602–10606 CrossRef CAS PubMed.
- S. E. Sloop, Patent No.: US 7.198.865 B2, 2007.
- M. Winter and J. O. Besenhard, Chem. Unserer Zeit, 1999, 33, 320–332 CrossRef CAS PubMed.
- R. Wagner, N. Preschitschek, S. Passerini, J. Leker and M. Winter, J. Appl. Electrochem., 2013, 43, 481–496 CrossRef CAS.
- K. Tasaki, A. Goldberg and M. Winter, Electrochim. Acta, 2011, 56, 10424–10435 CrossRef CAS PubMed.
- H. J. Santner, C. Korepp, M. Winter, J. O. Besenhard and K. C. Möller, Anal. Bioanal. Chem., 2004, 379, 266–271 CrossRef CAS PubMed.
- C. Korepp, H. J. Santner, T. Fujii, M. Ue, J. O. Besenhard, K. C. Möller and M. Winter, J. Power Sources, 2006, 158, 578–582 CrossRef CAS PubMed.
- S. Dalavi, M. Xu, B. Ravdel, L. Zhou and B. L. Lucht, J. Electrochem. Soc., 2010, 157, A1113–A1120 CrossRef CAS PubMed.
- M. Winter, Z. Phys. Chem., 2009, 223, 1395 CrossRef CAS.
- D. Aurbach, M. L. Daroux, P. W. Faguy and E. Yeager, J. Electrochem. Soc., 1987, 134, 1611–1620 CrossRef CAS PubMed.
- E. Peled, D. Golodnitsky, G. Ardel and V. Eshkenazy, Electrochim. Acta, 1995, 40, 2197–2204 CrossRef CAS.
- E. Peled, D. Golodnitsky and G. Ardel, J. Electrochem. Soc., 1997, 144, L208–L210 CrossRef CAS PubMed.
- H. Bryngelsson, M. Stjerndahl, T. Gustafsson and K. Edström, J. Power Sources, 2007, 174, 970–975 CrossRef CAS PubMed.
- P. Verma, P. Maire and P. Novák, Electrochim. Acta, 2010, 55, 6332–6341 CrossRef CAS PubMed.
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS PubMed.
- H. Yoshida, T. Fukunaga, T. Hazama, M. Terasaki, M. Mizutani and M. Yamachi, J. Power Sources, 1997, 68, 311–315 CrossRef CAS.
- G. E. Blomgren, J. Power Sources, 1999, 81–82, 112–118 CrossRef CAS.
- U. Heider, R. Oesten and M. Jungnitz, J. Power Sources, 1999, 81–82, 119–122 CrossRef CAS.
- X. Zhang, P. N. Ross, R. Kostecki, F. Kong, S. Sloop, J. B. Kerr, K. Striebel, E. J. Cairns and F. McLarnon, J. Electrochem. Soc., 2001, 148, A463–A470 CrossRef CAS PubMed.
- K. Tasaki, K. Kanda, S. Nakamura and M. Ue, J. Electrochem. Soc., 2003, 150, A1628–A1636 CrossRef CAS PubMed.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327–A2334 CrossRef CAS PubMed.
- T. Sasaki, T. Abe, Y. Iriyama, M. Inaba and Z. Ogumi, J. Power Sources, 2005, 150, 208–215 CrossRef CAS PubMed.
- T. Sasaki, T. Abe, Y. Iriyama, M. Inaba and Z. Ogumi, J. Electrochem. Soc., 2005, 152, A2046–A2050 CrossRef CAS PubMed.
- L. Gireaud, S. Grugeon, S. Pilard, P. Guenot, J.-M. Tarascon and S. Laruelle, Anal. Chem., 2006, 78, 3688–3698 CrossRef CAS PubMed.
- G. Gachot, S. Grugeon, M. Armand, S. Pilard, P. Guenot, J.-M. Tarascon and S. Laruelle, J. Power Sources, 2008, 178, 409–421 CrossRef CAS PubMed.
- G. Gachot, P. Ribière, D. Mathiron, S. Grugeon, M. Armand, J.-B. Leriche, S. Pilard and S. Laruelle, Anal. Chem., 2011, 83, 478–485 CrossRef CAS PubMed.
- L. Terborg, S. Nowak, S. Passerini, M. Winter, U. Karst, P. R. Haddad and P. N. Nesterenko, Anal. Chim. Acta, 2012, 714, 121–126 CrossRef CAS PubMed.
- L. Terborg, S. Weber, F. Blaske, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2013, 242, 832–837 CrossRef CAS PubMed.
- B. Vortmann, S. Nowak and C. Engelhard, Anal. Chem., 2013, 85, 3433–3438 CrossRef CAS PubMed.
- L. Terborg, S. Weber, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2014, 245, 836–840 CrossRef CAS PubMed.
- M. Grützke, V. Kraft, W. Weber, C. Wendt, A. Friesen, S. Klamor, M. Winter and S. Nowak, J. Supercrit. Fluids, 2014, 94, 216–222 CrossRef PubMed.
- V. Kraft, M. Grützke, W. Weber, M. Winter and S. Nowak, J. Chromatogr. A, 2014, 1354, 92–100 CrossRef CAS PubMed.
- M. Grützke, V. Kraft, B. Hoffmann, S. Klamor, J. Diekmann, A. Kwade, M. Winter and S. Nowak, J. Power Sources, 2015, 273, 83–88 CrossRef PubMed.
- C. Schultz, V. Kraft, M. Pyschik, S. Weber, F. Schappacher, M. Winter and S. Nowak, J. Electrochem. Soc., 2015, 162, A629–A634 CrossRef CAS PubMed.
- W. Weber, V. Kraft, M. Grützke, M. Winter and S. Nowak, J. Chromatogr. A, 2015, 1394, 128–136 CrossRef CAS PubMed.
- A. W. Golubkov, D. Fuchs, J. Wagner, H. Wiltsche, C. Stangl, G. Fauler, G. Voitic, A. Thaler and V. Hacker, RSC Adv., 2014, 4, 3633–3642 RSC.
- Y. Fu, S. Lu, K. Li, C. Liu, X. Cheng and H. Zhang, J. Power Sources, 2015, 273, 216–222 CrossRef CAS PubMed.
- M. Reichert, J. Haetge, D. Berghus, C. Wendt, V. Meier, U. Rodehorst, S. Passerini, F. Schappacher and M. Winter, ECS Trans., 2014, 61, 87–103 CrossRef PubMed.
- D. Aurbach, B. Markovsky, A. Rodkin, M. Cojocaru, E. Levi and H.-J. Kim, Electrochim. Acta, 2002, 47, 1899–1911 CrossRef CAS.
- T. A. Immel, M. Grützke, A.-K. Späte, U. Groth, P. Öhlschlager and T. Huhn, Chem. Commun., 2012, 48, 5790–5792 RSC.
- P. W. Atkins and J. d. Paula, Kurzlehrbuch Physikalische Chemie, 2005 Search PubMed.
- S. E. Sloop, J. K. Pugh, S. Wang, J. B. Kerr and K. Kinoshita, Electrochem. Solid-State Lett., 2001, 4, A42–A44 CrossRef CAS PubMed.
- S. Castillo, F. Ansart, C. Laberty-Robert and J. Portal, J. Power Sources, 2002, 112, 247–254 CrossRef CAS.
- M. J. Lain, J. Power Sources, 2001, 97–98, 736–738 CrossRef CAS.
- S. Krüger, C. Hanisch, A. Kwade, M. Winter and S. Nowak, J. Electroanal. Chem., 2014, 726, 91–96 CrossRef PubMed.
- Y. Wang, Z.-S. Feng, J.-J. Chen and C. Zhang, Mater. Lett., 2012, 71, 54 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |