
Selective and sensitive SERS sensor for detection of Hg2+ in environmental water base on rhodamine-bonded and amino group functionalized SiO2-coated Au–Ag core–shell nanorods†
Abstract
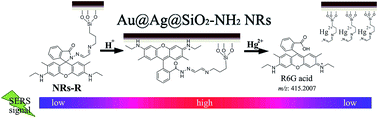
Here, we designed a new type of SERS sensor with high selectivity and sensitivity for the determination of Hg2+ at picomolar concentrations based on hydrolysis reactions. The Rhodamine 6G-derived Schiff base bonded and amino group functionalized SiO2-coated Au–Ag core–shell nanorods (Au@Ag@SiO2–NH2-R6G, NR-Rs), which show extraordinary enhancement factors (EF) and good stability, were employed as the SERS substrate of the sensor. The tunable ability of the chemical bonding of the substrate generates appropriate probe molecules bound to the surface of the nanorods (NRs) and improves the selectivity and sensitivity of the SERS sensor dramatically. The limit of detection (LOD) of Hg2+ obtained using the present SERS sensor was 0.33 pmol L−1. Furthermore, the proposed substrate has pH-responsive ability. The present sensor is suitable for the on-site determination of pH and Hg2+ by combining with a portable Raman spectrometer.
- This article is part of the themed collection: Surface enhanced Raman Spectroscopy: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

