
Development of magnetoelectric CoFe2O4 /poly(vinylidene fluoride) microspheres
R. Gonçalvesab,
P. Martins*a,
D. M. Correiaab,
V. Sencadasa,
J. L. Vilasc,
L. M. Leóncd,
G. Botelhob and
S. Lanceros-Méndez*a
aCentro/Departamento de Física, Universidade do Minho, 4710-057 Braga, Portugal. E-mail: pmartins@fisica.uminho.pt; lanceros@fisica.uminho.pt
bCentro/Departamento de Química, Universidade do Minho, 4710-057 Braga, Portugal
cDepartamento de Química Física, Facultad de Ciencia y Tecnología, Universidad del País Vasco/EHU, Apdo.644, Bilbao E-48080, Spain
dBasque Center for Materials, Applications and Nanostructures (BCMaterials), Parque Tecnológico de Bizkaia, Ed. 500, Derio 48160, Spain
First published on 14th April 2015
Abstract
Magnetoelectric microspheres based on piezoelectric poly(vinylidene fluoride) (PVDF) and magnetostrictive CoFe2O4 (CFO), a novel morphology for polymer-based ME materials, have been developed by an electrospray process. The CFO nanoparticle content in the (3–7 μm diameter) microspheres reaches values up to 27 wt%, despite their concentration in the starting solution reaching values up to 70 wt%. Additionally, the inclusion of magnetostrictive nanoparticles into the polymer spheres has no relevant effect on the piezoelectric β-phase content (≈60%), crystallinity (40%) and the onset degradation temperature (460–465 °C) of the polymer matrix. The multiferroic microspheres show a maximum piezoelectric response |d33| ≈ 30 pC N−1, leading to a magnetoelectric response of Δ|d33| ≈ 5 pC N−1 obtained when a 220 mT DC magnetic field was applied. It is also shown that the interface between CFO nanoparticles and PVDF (from 0 to 55%) has a strong influence on the ME response of the microspheres. The simplicity and the scalability of the processing method suggest a large application potential of this novel magnetoelectric geometry in areas such as tissue engineering, sensors and actuators.
Introduction
The magnetoelectric (ME) effect, defined as the variation of the electric polarization in response to an applied magnetic field or the variation of the magnetization under an applied electrical field is a scientifically interesting and technological useful phenomenon with an increasing range of applications in areas such as computer memories, smart sensors, actuators, high frequency microelectronic devices and biomedical materials.1–4 The ME effect can occur on single-phase materials or in composites due to the combination of magnetostrictive and piezoelectric responses.5–8 Single-phase ME materials, typically show very low ME coupling exhibited at low temperatures, hindering their implementation into technological applications.1,7,9 Multiferroic composites emerged as an interesting possibility for device applications as in those composites, consisting on the combination of magnetostrictive and piezoelectric phases, the ME effect is the result of a product property, i.e., the mechanical deformation induced by a magnetic field due to the magnetostriction of one of the phases, results in a dielectric polarization variation due to the piezoelectric effect of the other phase, allowing large ME effects at room temperature.2,7,10ME composite materials can be ceramic or polymer based. Ceramic-based ME materials exhibit ME coefficients three orders of magnitude higher than polymer-based ME materials, but they are limited by reactions at the interface regions which lead to high dielectric losses, hindering sustainable device applications.1
Thus, polymer-based ME materials have attracted increasing interest from the industry since they solve the abovementioned application problems.1,11 Further, in polymer-based ME materials strain coupling does not deteriorate with operation, as the magnetostrictive material is in direct contact with and completely surrounded by the piezoelectric polymer matrix, they show simple and scalable production methods, a flexible structure without large leakage currents, can be fabricated by conventional low-temperature polymer processing into a variety of forms, such as thin sheets or molded shapes, can exhibit tailored mechanical properties, flexibility, lightweight, versatility, low cost and even biocompatibility.1,2
In particular, polymer-based ME spheres composed by magnetostrictive nanoparticles within a piezoelectric polymer matrix, can open new applications areas and solve some drawbacks of the traditional polymer-based structures (nanocomposites, polymer as a binder and laminates) such as agglomeration, irregular distributions and the difficulty to shape in a miniaturized form.1,12
Polymer-based micro and nanospheres undergo an increasing demand and applicability as biomaterials for cell culture, drug delivery systems, electro-optic and luminescent devices, heterogeneous catalysis and polymer powder impregnation of inorganic fibers in composites.13–16
Particularly, low-scale piezoelectric materials such as spheres show strong potentials for improved energy harvesters with higher volume efficiency, nano-sensors and nano-actuators and nano-mats guiding cell distribution.17,18 The addition of magnetostrictive materials into the piezoelectric spheres allows the use of the resulting composite also as magnetic nano-sensors and actuators, as well as to take advantage of the induced ME phenomenon.7
To our knowledge there are no previous reports on polymer-based ME spheres, that can be an innovative and desired solution for applications in which multifunctional active response is needed (either magnetic to electrical or mechanical to electrical responses, due to the ME and piezoelectric effects) such as in non-invasive control of cell growth and differentiation, active drug release and tissue stimulation.14,16
For the formation of polymer microspheres several methods have been used such as gas atomization, microdroplet, dispersion polymerization, evaporation and precipitation, emulsion polymerization,13 oil in water (O/W) or water in oil (W/O) emulsions, coacervation and spray drying, among others.19 Unlike previous methods that require high-energy input devices like sonicators and/or high-cost devices such as high-pressure homogenisers, electrospray technique is a straightforward and versatile technique featuring advantages like ambient condition and single-step processing, high reproducibility, high yield and economical set-up.15,20,21
In this work the development of novel CoFe2O4 (CFO)/polyvinylidene fluoride (PVDF) multiferroic spheres is reported, with large potential applications in the biomedical, sensing, actuation, catalysis and energy fields.13–15
PVDF, is a polymer with five possible distinct crystalline phases named as α, β-phase, γ, δ and ε. The β-phase shows the strongest piezoelectric response and is thus required as the piezoelectric component around the magnetostrictive particles. PVDF in the β-phase shows thus biocompatibility, high piezoelectric response, large chemical and radiation resistance, easy shaping and low cost.11,22–24 CFO nanoparticles will be embedded within the piezoelectric polymer sphere matrix and were selected as the magnetostrictive phase due to their chemical stability, mechanical hardness, wear resistance, ease of synthesis, large magnetostriction, high Curie temperature, low cost and simple processability.25,26 Additionally, the high magneto-crystalline anisotropy of the CFO nanoparticles is very interesting and useful for their use in medical applications.27
Experimental
Materials and methods
Poly(vinylidene fluoride), PVDF, reference Solef 1010, was acquired from Solvay. Analytical grade tetrahydrofuran (THF) and N,N-dimethyl formamide (DMF) were purchased from Panreac and Merck, respectively. CoFe2O4, CFO, nanoparticles with 35–55 nm particle size, were purchased from Nanoamor. Laboratory grade Triton X-100 was purchased from Sigma-Aldrich.Composite preparation
The CFO nanoparticles were dispersed in DMF solvent and Triton X-100 in an ultrasound bath during 4 h to ensure good dispersion and avoid nanoparticle agglomeration. Then, PVDF and THF were added and placed in a Teflon mechanical stirrer and an ultrasound bath until complete dissolution of the polymer. Composite solutions with CFO contents between 10 weight percentage (wt%) and 70 wt% were produced.Electrospray processing
The composite solution was placed in a commercial plastic syringe fitted with a steel needle with inner diameter of 0.5 mm. Electrospray was conducted by applying 20 kV with a PS/FC30P04 power source from Glassman. A syringe pump (Syringepump) feed the polymer solution into the tip at a 1 mL h−1 rate. The electrosprayed samples were collected on a grounded collecting plate placed at 20 cm from the needle tip.Sample characterization
The morphology of the CFO/PVDF spheres was evaluated by Scanning Electron Microscopy (SEM) (Quanta 650, from FEI) with an accelerating voltage of 5 kV. Sphere average diameter and distribution was calculated over approximately 30 microspheres using SEM images (5000× magnification) and the ImageJ software.The magnetic properties of the multiferroic spheres were evaluated by measuring the magnetization loops M(H) up to 10 kOe using an Oxford Instruments vibrating sample magnetometer. Fourier Transform Infrared Spectroscopy (FTIR) technique was carried out at room temperature in a Bruker alpha apparatus in ATR mode from 4000 to 400 cm−1 using 24 scans at a resolution of 4 cm−1. Specific bands such as the ones at 766 and 840 cm−1 have been identified to correspond to the α and β-phase, respectively, allowing the calculation of the polymer phase content after the procedure described in.22 The β-phase fraction (F(β)) can thus be determined by applying eqn (1):
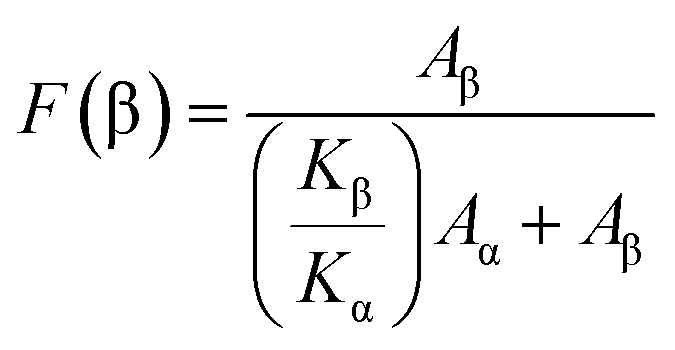 | (1) |
The thermal behaviour of the samples was determined by Differential Scanning Calorimetry (DSC), measurements in a Mettler Toledo 822e apparatus with sample robot and STAR software, using a heating rate of 10 °C min−1 under nitrogen purge (50 mL min−1); and by ThermoGravimetric Analysis (TGA). For the TGA measurements, samples were transferred to open ceramic crucibles with capacity of 60 μL and analysed using a METTLER TGA/SDTA 851 thermobalance operating between 200 °C and 700 °C. A heating rate of 10 ± 0.2 °C min−1 and nitrogen flow rate of 50 mL min−1 were used.
The crystallinity content (Xc) of the PVDF samples was calculated applying eqn (2):21
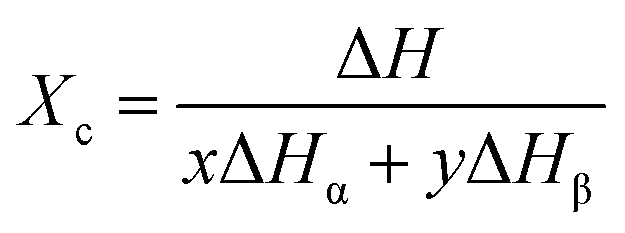 | (2) |
Form the TGA results, the nanofiller/polymer interface region of the CFO/PVDF spheres was obtained, applying eqn (3):23
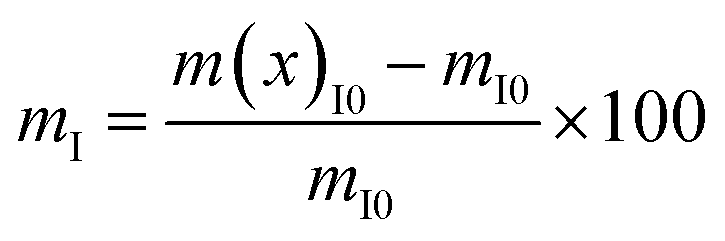 | (3) |
After poling conditions optimization, 30 min of corona poling at 10 kV and 120 °C were applied in a home-made chamber in order to optimize the piezoelectric response of the samples. Then, the piezoelectric response (d33) of the samples was analysed with a wide range d33-meter (model 8000, APC Int Ltd).
The ME character of the CFO/PVDF spheres was evaluated by the difference in the piezoelectric response obtained with and without the application of a 220 Oe DC magnetic field (Δd33).
Results and discussion
VSM technique has proved to be a precise technique able to accurately determine the magnetic nanoparticle content on composites.29–31 Thus, the hysteresis curves shown in Fig. 1 were first used to evaluate the efficiency of the particle loading process, i.e. the relation between the content of the CFO nanoparticles within the solution and the concentration in the obtained spheres (Fig. 1).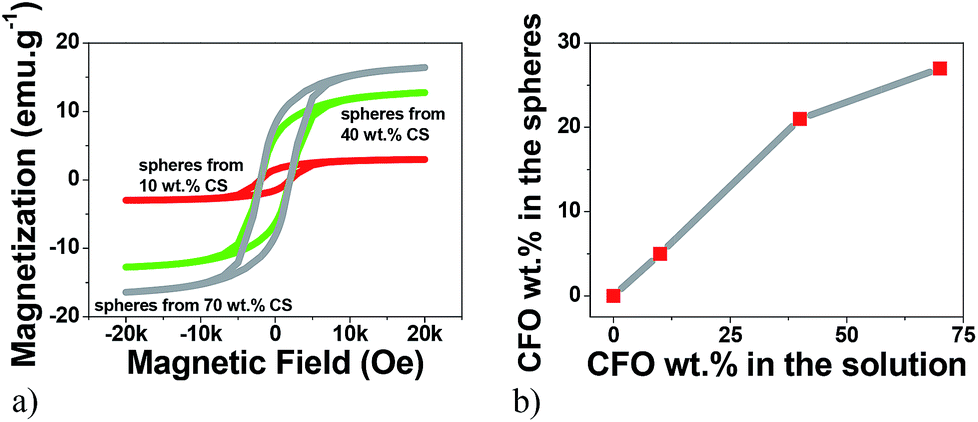 | ||
| Fig. 1 (a) Room temperature hysteresis loops for the multiferroic CFO/PVDF spheres. (b) Relation between the wt% of CFO nanoparticles within the solution and the wt% of CFO nanoparticles within the multiferroic spheres, obtained from the hysteresis loops. | ||
Fig. 1a reveals the typical ferromagnetic behavior of the CFO/PVDF spheres. For all compositions, the magnetization saturates at ≈2 kOe. As expected, the magnetization saturation increases with increasing nanoparticle filler content. By comparing the saturation magnetization value of the pure CFO nanoparticles with the ones from Fig. 1a it is possible to determine, through eqn (4), the precise amount of CFO nanoparticles within the multiferroic sphere (Table 1).
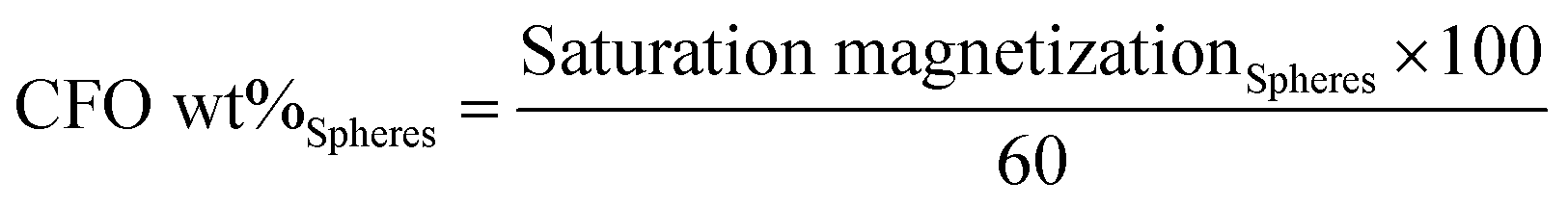 | (4) |
Fig. 1b and Table 1 show that with 10 wt%, 40 wt% and 70 wt% CFO content within the solution leads to spheres with 5 wt%, 21 wt% and 27 wt% CFO contents, respectively. Thus, the maximum CFO content allowed in the spheres starts to saturate at ∼20 wt%, since an increase of 30 wt% in the solution wt% content of CFO (from 40% to 70%) leads to just an increase of just 6% of CFO nanoparticles inside the multiferroic sphere (from 21% to 27%).
In this way, the concentrations of CFO nanoparticles in the electrosprayed spheres is lower than the ones on the composite solutions, in agreement to previous reports32 and can be attributed to the higher density of the CFO nanoparticles (when compared to the polymer matrix) that causes the settling of some nanoparticles on the bottom of the syringe during the electrospinning process. Further, some contributions can also come from a partial blockage of the needle hole by agglomeration of nanoparticles, due to the flow funnelling towards the needle. Fig. 2a–e shows representative SEM images of ME CFO/PVDF spheres with 5–27 wt% ferrite content.
 | ||
| Fig. 2 Morphology of PVDF polymer (a and b) and the multiferroic CFO/PVDF microspheres with CFO wt% 5 (c), 21 (d) and 27 (e) CFO nanoparticle content. | ||
The low magnification image (Fig. 2a) shows a homogeneous production of multiferroic spheres, with good dispersion and spherical shape. Spheres diameters were between 3 and 7 μm, nearly independently of the CFO filler content. The insertion of the CFO magnetic fillers within the PVDF polymer sphere originates just a slight decrease of the average sphere diameter.
Backscattering images (Fig. 2c–e) reveal that the CFO nanoparticles are effectively inside (white zones of Fig. 2c–e) the polymer spheres, wrapped by the polymer matrix (spherical structure of Fig. 2b), giving rise to the desired multiferroic polymer composite structure.
Since the presence of the piezoelectric β crystalline phase of PVDF is an essential requirement to obtain ME response on PVDF based ME materials,1 FTIR was used to identify and quantify the β-phase content of PVDF.
For the pure polymer and the CFO/PVDF microspheres, typical FTIR spectra are presented in Fig. 3a and the calculated F(β), eqn (1), is represented in Fig. 3b.
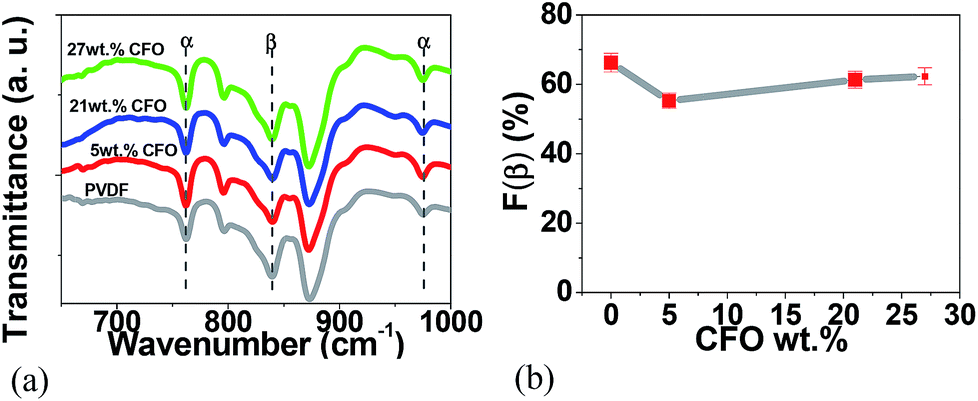 | ||
| Fig. 3 FTIR spectra of (a) pure PVDF microspheres and CFO/PVDF composites microspheres with 5, 21 and 27 wt% filler content and (b) variation of β-phase content as a function of CFO content. | ||
Fig. 3a shows that the crystalline phases of the polymer matrix in the microspheres are mainly in the β-phase and no significant differences between the spectra of the different composite microsphere are detected. All microspheres, pure PVDF and CFO/PVDF composites, show β-phase contents between 65 and 75% and this value is independent of the CFO content. It is to notice that those β-phase contents are compatible with the maximum piezoelectric response of the polymer, as it has been verified in.33 In this way, the β-phase formation is mainly attributed to the low solvent evaporation temperature (≤60 °C), which mainly leads to the crystallization of the polymer in this phase.15,34 Further, electrostatic interaction of the filler nanoparticles with the highly polar polymer chains certainly reinforce this effect, as it has been verified in samples prepared after melting, that are nucleated in the β-phase, whereas the polymer without fillers remain in the α-phase.15
Fig. 4a shows the DSC thermograph of the microspheres of PVDF and CFO/PVDF composites. From the melting endotherm and applying eqn (2), the degree of crystallinity (Xc) was obtained, as represented in Fig. 4b.
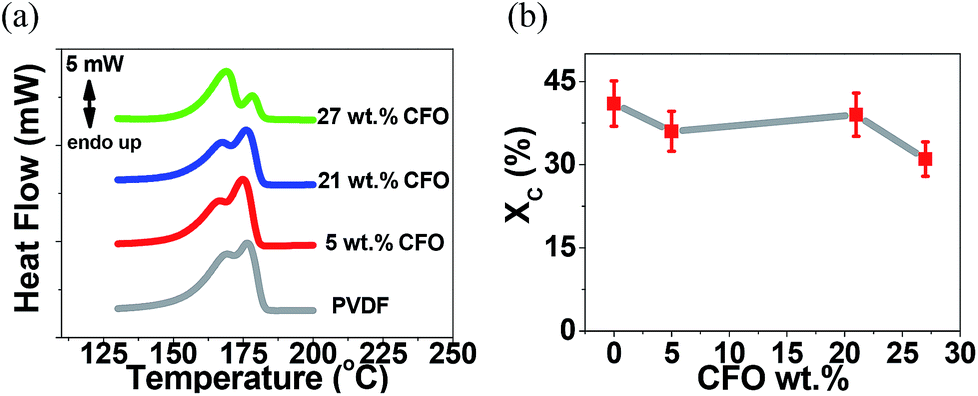 | ||
| Fig. 4 (a) DSC thermographs and (b) degree of crystallinity of the pristine PVDF and the CFO/PVDF composite microspheres. | ||
The DSC thermographs of all samples are characterized by a double endothermic peak, related to the polymer crystallization in both α and β crystalline structures as confirmed by FTIR (Fig. 3) results35 and the presence of the nanofillers. In both cases ill-crystallized region arises with lower melting temperature due to the larger energy of the imperfect structures. The degree of crystallinity values (Fig. 4b) are in good agreement with the ones obtained in PVDF processed by similar procedures.36 Additionally, the overall lower degree of crystallinity is slightly lower when the fillers are present in the polymer matrix, which is attributed to hindered crystallization due to the presence of the fillers, which can act as nucleation centers for crystallization, but also hinder spherulite growth.31,35
The interface between magnetostrictive materials and piezoelectric polymers is one of the most sensitive parameters influencing the ME response of the composites. This interface can be determined by the TGA results presented in (Fig. 5).23,36
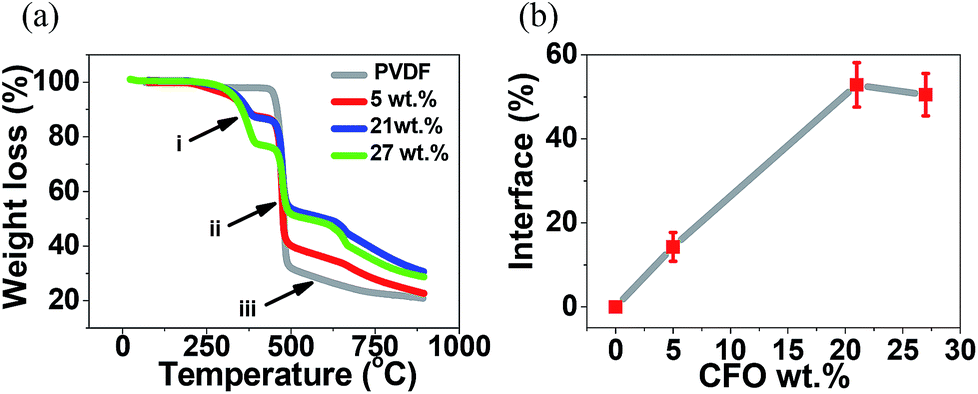 | ||
| Fig. 5 (a) TGA thermographs for the different samples and (b) interface volume between nanoparticles and polymer as a function of CFO nanoparticle concentration. | ||
In all composite spheres samples, with and without CFO nanoparticles, the typical two step thermal degradation, characteristic of PVDF, was observed.37 The onset temperature, defined as the temperature at which the polymer lost 1% of its weight, was found to be ≈460 °C for the pure PVDF microspheres, slightly lower than the ones obtained on CFO/PVDF microspheres that was around ≈465 °C. These results shows that the addition of the CFO nanoparticles into the PVDF spheres slightly improves the thermal stability of the microspheres. Such effect has already been reported in previous studies23 and can be attributed to two factors: (a) the CFO filler in the composite can hinder the formation and escape of volatile by-products during heating and (b) the thermal motion of PVDF segments near the CFO surfaces may be restricted because of the physical interlock and electrostatic interaction.38
The first degradation step occurs between ≈400 and ≈500 °C (ii), being the polymer maximum degradation temperature not influenced by the CFO content. In this initial degradation step the decomposition mechanism is chain-stripping where carbon–hydrogen and carbon–fluorine scission occurs, the presence of both hydrogen and fluorine radicals leading to the formation of hydrogen fluoride.37,39
The second degradation step occurs between ≈500 and ≈850 °C (iii), and the differences observed in the plots relatively to the pure PVDF spheres sample are to be ascribed to the presence of CFO nanoparticles, as the different phases of PVDF show similar thermal degradation behavior.40 This second step is a complex degradation process resulting in poly(aromatization). The polyenic sequence formed previously on the first degradation step is unstable and, as a consequence, the macromolecules formed undergo further reactions leading to scission followed by the formation of new aromatic molecules.23,37,41
Previously to these typical two thermal degradation steps, an additionally degradation was observed between ≈290 and≈ 400 °C (a) resultant from the degradation of the Triton X-100.42
Fig. 5b shows the mass fraction of the polymer located at the interface as a function of the CFO content, calculated after eqn (3). The interface value increases with increasing ferrite loading as a result of the increased number of particles interacting with the polymer matrix up to a filler content of ∼20 wt%, after this value, increasing CFO content has a result a small decrease in the CFO/PVDF interface, explained by the fact that a larger filler content can lead to the formation of clusters and agglomerations and therefore a decrease of the overall surface contact area. The highest interface value (55%) was obtained for the CFO/PVDF spheres with 21 wt% ferrite content. This interface value is ≈40% higher than the one reported to CFO/PVDF multiferroic composite films43 and will lead to an increased ME coupling due to the larger contact area between the magnetostrictive and piezoelectric phases.
The ME coupling was measured44,45 by evaluating the piezoelectric response of the composites without and with an applied magnetic field of 220 mT (Fig. 6).
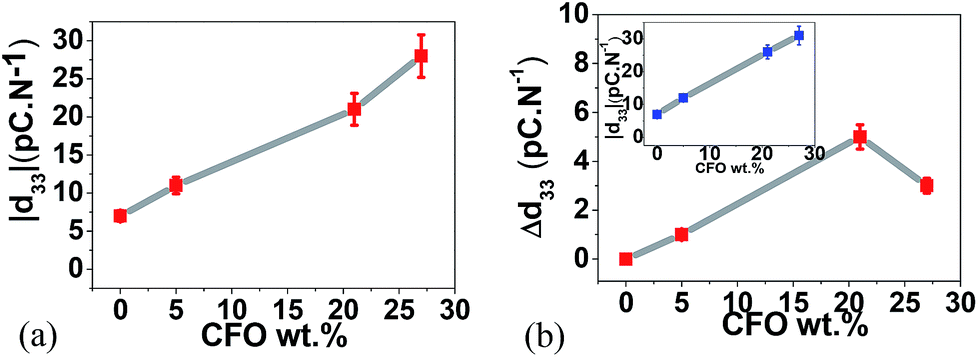 | ||
| Fig. 6 (a) Modulus of the piezoelectric coefficient |d33| as a function of CFO wt% and (b) |d33| (inset) and Δ|d33| as a function of CFO wt% with the applied 220 mT DC magnetic field. | ||
Fig. 6a represents the variation of the piezoelectric response of the samples (polymer films made out of spheres-Fig. 2) as a function of filler content. The presence of the CFO nanoparticles improves the piezoelectric response of the composite spheres due to the strong interfacial interactions between particles and polymer.31,34,46 With higher ferrite contents, the interfacial elastic effect is stronger and leads to higher piezoelectric responses.46
Once a 220 mT DC magnetic field was applied with two permanent magnets, at the same time that the piezoelectric response is being measured, an increase in the |d33| value is observed in the composite samples (Fig. 6b, inset) but no variation is detected in the pristine polymer samples revealing the ME character of the multiferroic spheres.
Since magnetostrictive CFO induce displacements at the interface between nanoparticles and polymer,47 with increasing interface value, a better coupling34 and interaction between the piezoelectric PVDF and the magnetostrictive CFO ferrites will be promoted, explaining the larger increase of the Δ|d33| with increasing nanoparticle content until ∼20 wt%. Such interaction will be hindered for higher CFO concentrations, due to the decrease of the previously shown interface area (Fig. 5b), leading to a lower ME coupling and a decrease in the |d33| variation value.
Conclusions
Magnetoelectric CFO/PVDF microspheres have been prepared by an electrospray process. The concentrations of CFO nanoparticles in the microspheres reaches values up to 0–27 wt%, though their concentration in solution reaches values up to 70 wt%. Spheres diameters were found to be between 3 and 7 μm, being the size nearly independent of the CFO filler content.The addition of CFO nanoparticles into the polymeric spheres has almost no effect on the β-phase content (≈60%), crystallinity (40%) and the onset degradation temperature (460–465 °C) of the polymer matrix.
The interface between CFO nanoparticles and PVDF was found to have a strong influence on the ME response of the CFO/PVDF spheres. Increased interface values (from 0 to 55%) had as result and optimized ME response (Δ|d33| from 0 to 5 pC N−1) when a 220 mT DC magnetic field was applied to the CFO/PVDF spheres with 21 wt% of ferrite. Thus, the overall properties of the ME microspheres, the simplicity and scalability of the processing method indicates a large potential of the CFO/PVDF multifunctional microspheres for developing advanced applications.
Acknowledgements
This work is funded by FEDER funds through the “Programa Operacional Factores de Competitividade – COMPETE” and by national funds from FCT – Portuguese Foundation for Science and Technology, in the framework of the strategic project Strategic Project PEST-C/FIS/UI607/2014 and PEst-C/QUI/UI0686/2014. Authors would like to thank to J. C. Dias for the TGA measurements. The authors also thank funding from “Matepro –Optimizing Materials and Processes, ref. NORTE-07-0124-FEDER-000037”, co-funded by the “Programa Operacional Regional do Norte” (ON.2 – O Novo Norte), under the “Quadro de Referência Estratégico Nacional” (QREN), through the “Fundo Europeu de Desenvolvimento Regional” (FEDER). P. Martins, R. Gonçalves, D. M. Correia and V. Sencadas acknowledges also support from FCT (SFRH/BPD/96227/2013, SFRH/BD/88397/2012, SFRH/BD/90215/2012, SFRH/BPD/64958/2009 grants respectively).Notes and references
- P. Martins and S. Lanceros-Méndez, Adv. Funct. Mater., 2013, 23, 3371–3385 CrossRef CAS PubMed.
- A. Maceiras, P. Martins, R. Gonçalves, G. Botelho, E. Venkata Ramana, S. K. Mendiratta, M. San Sebastián, J. L. Vilas, S. Lanceros-Mendez and L. M. León, Eur. Polym. J., 2015, 64, 224–228 CrossRef CAS PubMed.
- J. Jin, F. Zhao, K. Han, M. A. Haque, L. Dong and Q. Wang, Adv. Funct. Mater., 2014, 24, 1067–1073 CrossRef CAS PubMed.
- W. Eerenstein, N. D. Mathur and J. F. Scott, Nature, 2006, 442, 759–765 CrossRef CAS PubMed.
- R. Gupta, M. Tomar, V. Gupta, Y. Zhou, A. Bhalla and S. Priya, Adv. Sci. Lett., 2014, 20, 1116–1119 CrossRef PubMed.
- P. Gupta and P. Poddar, RSC Adv., 2015, 5, 10094–10101 RSC.
- A. Baji, Y. W. Mai, R. Yimnirun and S. Unruan, RSC Adv., 2014, 4, 55217–55223 RSC.
- N. A. Spaldin and M. Fiebig, Science, 2005, 309, 391–392 CrossRef CAS PubMed.
- K. Chand Verma, S. K. Tripathi and R. K. Kotnala, RSC Adv., 2014, 4, 60234–60242 RSC.
- M. Fiebig, J. Phys. D: Appl. Phys., 2005, 38, R123–R152 CrossRef CAS.
- P. Martins, C. M. Costa, J. C. C. Ferreira and S. Lanceros-Mendez, J. Phys. Chem. B, 2012, 116, 794–801 CrossRef CAS PubMed.
- A. Chaudhuri and K. Mandal, J. Magn. Magn. Mater., 2015, 377, 441–445 CrossRef CAS PubMed.
- J. Otaigbe, M. Barnes, K. Fukui, B. Sumpter and D. Noid, in Polymer Physics and Engineering, Springer, Berlin, Heidelberg, 2001, vol. 154, pp. 1–86 Search PubMed.
- K. Yue, R. Guduru, J. Hong, P. Liang, M. Nair and S. Khizroev, PLoS One, 2012, 7, 44040 Search PubMed.
- C. Luo, T. Okubo, M. Nangrejo and M. Edirisinghe, Polym. Int., 2015, 64, 183–187 CrossRef CAS PubMed.
- Y. Cao, B. Wang, Y. Wang and D. Lou, RSC Adv., 2014, 4, 30430–30439 RSC.
- H. B. Kang, C. S. Han, J. C. Pyun, W. H. Ryu, C. Y. Kang and Y. S. Cho, Compos. Sci. Technol., 2015, 111, 1–8 CrossRef CAS PubMed.
- S. Banerjee, D. Konwar and A. Kumar, Sens. Actuators, B, 2014, 190, 199–207 CrossRef CAS PubMed.
- Y.-H. Lee, F. Mei, M.-Y. Bai, S. Zhao and D.-R. Chen, J. Controlled Release, 2010, 145, 58–65 CrossRef CAS PubMed.
- L. Konermann, R. G. McAllister and H. Metwally, J. Phys. Chem. B, 2014, 118, 12025–12033 CrossRef CAS PubMed.
- D. M. Correia, R. Gonçalves, C. Ribeiro, V. Sencadas, G. Botelho, J. L. G. Ribelles and S. Lanceros-Méndez, RSC Adv., 2014, 4, 33013–33021 RSC.
- P. Martins, A. C. Lopes and S. Lanceros-Mendez, Prog. Polym. Sci., 2014, 39, 683–706 CrossRef CAS PubMed.
- R. Gonçalves, P. M. Martins, C. Caparrós, P. Martins, M. Benelmekki, G. Botelho, S. Lanceros-Mendez, A. Lasheras, J. Gutiérrez and J. M. Barandiarán, J. Non-Cryst. Solids, 2013, 361, 93–99 CrossRef PubMed.
- T. Wu, B. Zhou, T. Zhu, J. Shi, Z. Xu, C. Hu and J. Wang, RSC Adv., 2015, 5, 7880–7889 RSC.
- K. Khaja Mohaideen and P. A. Joy, J. Magn. Magn. Mater., 2013, 346, 96–102 CrossRef CAS PubMed.
- J. Wang, Z. Zhou, L. Wang, J. Wei, H. Yang, S. Yang and J. Zhao, RSC Adv., 2015, 5, 7349–7355 RSC.
- S. Amiri and H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 1–8 CrossRef CAS PubMed.
- L. L. Sun, B. Li, Z. G. Zhang and W. H. Zhong, Eur. Polym. J., 2010, 46, 2112–2119 CrossRef CAS PubMed.
- W. Lin, Y. Miao, H. Zhang, B. Liu, Y. Liu, B. Song and J. Wu, J. Lightwave Technol., 2013, 31, 2599–2605 CrossRef.
- M. F. Valan, A. Manikandan and S. A. Antony, J. Nanosci. Nanotechnol., 2015, 15, 4543–4551 CrossRef CAS PubMed.
- P. Martins, A. Lasheras, J. Gutierrez, J. M. Barandiaran, I. Orue and S. Lanceros-Mendez, J. Phys. D: Appl. Phys., 2011, 44, 495303 CrossRef.
- X. Chen, S. Wei, C. Gunesoglu, J. Zhu, C. S. Southworth, L. Sun, A. B. Karki, D. P. Young and Z. Guo, Macromol. Chem. Phys., 2010, 211, 1775–1783 CrossRef CAS PubMed.
- J. Gomes, J. S. Nunes, V. Sencadas and S. Lanceros-Mendez, Smart Mater. Struct., 2010, 19, 065010 CrossRef.
- P. Martins, C. M. Costa, M. Benelmekki, G. Botelho and S. Lanceros-Mendez, CrystEngComm, 2012, 14, 2807–2811 RSC.
- C. Merlini, G. M. O. Barra, T. Medeiros Araujo and A. Pegoretti, RSC Adv., 2014, 4, 15749–15758 RSC.
- R. Belouadah, D. Guyomar, B. Guiffard and J.-W. Zhang, Phys. B, 2011, 406, 2821–2826 CrossRef CAS PubMed.
- G. Botelho, S. Lanceros-Mendez, A. M. Gonçalves, V. Sencadas and J. G. Rocha, J. Non-Cryst. Solids, 2008, 354, 72–78 CrossRef CAS PubMed.
- L. Fang, W. Wu, X. Huang, J. He and P. Jiang, Compos. Sci. Technol., 2015, 107, 67–74 CrossRef CAS PubMed.
- S. Zulfiqar, M. Rizvi and A. Munir, Polym. Degrad. Stab., 1994, 46, 19–23 CrossRef CAS.
- M. P. Silva, V. Sencadas, G. Botelho, A. V. Machado, A. G. Rolo, J. G. Rocha and S. Lanceros-Mendez, Mater. Chem. Phys., 2010, 122, 87–92 CrossRef CAS PubMed.
- M. L. O'Shea, C. Morterra and M. J. D. Low, Mater. Chem. Phys., 1990, 26, 193–209 CrossRef.
- K. Mitsuda, H. Kimura and T. Murahashi, J. Mater. Sci., 1989, 24, 413–419 CrossRef CAS.
- P. Martins, C. M. Costa, M. Benelmekki, G. Botelho and S. Lanceros-Méndez, J. Mater. Sci., 2013, 48, 2681–2689 CrossRef CAS.
- M. Vopsaroiu, M. Stewart, T. Hegarty, A. Muniz-Piniella, N. McCartney, M. Cain and G. Srinivasan, Meas. Sci. Technol., 2008, 19, 045106 CrossRef.
- S. H. Xie, Y. Y. Liu and J. Y. Li, Front. Phys., 2012, 7, 399–407 CrossRef PubMed.
- X. Liu, S. Liu, M. G. Han, L. Zhao, H. Deng, J. Li, Y. Zhu, L. Krusin-Elbaum and S. O'Brien, Nanoscale Res. Lett., 2013, 8(1), 374 CrossRef PubMed.
- J. M. Rondinelli, M. Stengel and N. A. Spaldin, Nat. Nanotechnol., 2008, 3, 46–50 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |