
New developments in direct functionalization of C–H and N–H bonds of purine bases via metal catalyzed cross-coupling reactions
Abstract
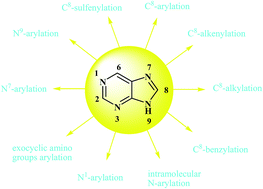
Purine bases have attracted much attention due to their potential biological activities. Developing more efficient methods for the modification of purine bases with a substitution such as aryl or alkyl is particularly interesting. This review gives an overview of new developments in direct functionalization of C–H and N–H bonds of purine bases via metal catalyzed cross-coupling reactions in recent years.
- This article is part of the themed collection: Organic chemistry collection
 Please wait while we load your content...
Please wait while we load your content...

