
Degradation of the azo dye Orange G in a fluidized bed reactor using iron oxide as a heterogeneous photo-Fenton catalyst
Abstract
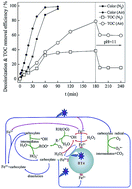
The heterogeneous Fenton process was employed to degrade Orange G (OG) using waste iron oxide as the catalyst in a three-phase fluidized bed reactor (3P-FBR). The morphology and the FTIR spectra of the used BT4 were compared with the fresh catalyst to illustrate the catalyst stability. The catalyst reusability was evaluated by measuring the colour removal in four successive cycles. The effects of major parameters, including pH, H2O2 concentration and catalyst addition on the decolourization of OG were investigated. A satisfactory decolourization efficiency (>92%) could be obtained under the conditions tested, and 78.9% of TOC removal was achieved at a 50 mg L−1 initial OG concentration, 25 mg L−1 H2O2 concentration, pH = 3 and 6 g L−1 BT4 addition. The decolourization of OG was mainly attributed to the homogeneous photo-Fenton reaction, while the heterogeneous catalytic process played an important role in TOC removal. The main intermediates were identified by the GC/MS technique and the degradation pathway of OG was proposed.
 Please wait while we load your content...
Please wait while we load your content...

