
Chemical interactions between zirconium and free oxide in molten fluorides
Miao Shena,
Hao Pengab,
Min Gea,
Chenyang Wanga,
Yong Zuoa and
Leidong Xie*a
aShanghai Institute of Applied Physics, Chinese Academy of Science, Shanghai 201800, P. R. China. E-mail: xieleidongsinap@163.com; Fax: +86-021-39194105; Tel: +86-021-39194105
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 29th April 2015
Abstract
The chemical interactions between zirconium and free oxide in FLiNaK melts at different zirconium to oxide (nZr/nO) molar ratios were studied by means of a carbothermal-reduction technique (LECO oxide analyzer) and Raman spectroscopy. ZrO2 precipitates were formed with nZr/nO ≤ 0.5, while ZrO2 was converted to Zr2OFx6−x complexes with nZr/nO > 0.5. The maximum amount of Zr2OFx6−x complexes in FLiNaK melts was found to be 0.020 mol kg−1. With an initial oxide concentration lower than 0.020 mol kg−1 and the required amount of ZrF4, the free oxide in FLiNaK melts could be completely converted to Zr2OFx6−x complexes, which would further prevent the formation of UO2 precipitates.
1. Introduction
The molten salt reactor (MSR) is one of the six internationally recognized Generation IV reactor concepts,1–3 which has been chosen as an ideal nuclear reactor to utilize uranium and thorium resources. Unlike other reactor types, MSR uses a 7LiF–BeF2 melt as a carrier salt, in which the fissile material (uranium in the form of 233UF4) and fertile material (thorium in the form of 232ThF4) are dissolved.4–6 UF4 is prone to react with free oxide (such as H2O and dissolved O2−), resulting UO2 with much higher melting point and lower solubility. Despite the loss of dissolved uranium, this may cause criticality concerns, as the large accumulation of UO2 would lead to superheated area. Therefore the oxide concentration should be strictly controlled.7–9 Early studies at Oak Ridge National Laboratory (ORNL) by Rosenthal et al.10 showed that the solubility product (KSP) of UO2 in LiF–BeF2 molten salt at 500 °C was 2.6 × 10−7 mol3 kg−3. Thus, the free oxide tolerance in the molten salt breeding reactor (MSBR) consisting of LiF–BeF2–ThF4–UF4 (71.7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16:12:0.3 mol%) is probably no greater than 30 ppm.
:16:12:0.3 mol%) is probably no greater than 30 ppm.
ORNL proposed two ways to prevent UO2 precipitation.11 One is to eliminate the free oxide by bubbling HF–H2 mixtures into the salt,11,12 as O2− ions react with an excess of HF according to the reaction O2− + 2HF → H2O↑ + 2F−. The other is to improve the oxide tolerance of uranium-based systems by adding some oxygen getters.10,13,14 Due to its high solubility in the fluorides,15,16 HF (g) retains stubbornly in the salts and causes the corrosion of structural material.17,18 Furthermore, HF–H2 purification is unable to confine the free oxide in fluorides to 30 ppm. Therefore, the preferable way is to introduce an oxygen getter to ensure the safe operation of MSR.
Five mole percent of ZrF4 was added to the LiF–BeF2–UF4 salts to getter any oxide impurities through the reaction 2H2O + ZrF4 = 4HF + ZrO2, as ZrF4 was more favorable to react with moisture than with UF4.8 Still, Korenko19 discovered that though ZrO2 were found in KF–LiF–NaF–UF4–ZrF4 by the X-ray diffraction (XRD) phase analysis, the UO2 could still be detected after the solidification of the molten system KF–LiF–NaF–UF4 without addition of ZrF4. Gibilaro20 also found the precipitation of ZrO2 and ZrO1.3F1.4 after CaO was added in the molten LiF–CaF2–ZrF4. These studies demonstrated that the zirconium-based systems reduced the free oxide by forming ZrO2 to prevent the precipitation of UO2. In this study, we found that the ZrO2 precipitates formed at nZr/nO ≤ 0.5, where nZr/nO refers to the molar ratios of zirconium to oxide. While with nZr/nO > 0.5, ZrO2 would be converted to Zr2OFx6−x complexes. Here, we primarily focused on the chemical interactions of zirconium and free oxide at different molar ratios and further explored the mechanism of ZrF4 protection against UO2 precipitation in molten FLiNaK.
2. Experimental section
2.1 Chemicals
The 300 g of LiF–NaF–KF [FLiNaK] mixture with a mole percent of 46.5:11.5:42 was weighed. The FLiNaK powders were heat to 300 °C and held at this temperature for 24 h, and then gradually heated to 600 °C under argon atmosphere. The salt was then dehydrated and deoxygenated by bubbling a mixture of HF (g)–H2 (g) (1:10 mole ratio). This process is consistent with that reported by ORNL-4616.11 After treatment, the oxide in FLiNaK melts can be lowered from 0.134 mol kg−1 to 0.008 mol kg−1 (Table 1), as detected by carbothermal-reduction technique using LECO RO-600 (LECO Co., Ltd.). Finally, this treated FLiNaK salt was transferred to a nickel crucible with an internal diameter of 78 mm. A certain amount of Li2O (Sigma-Aldrich, 99.99%) was added into FLiNaK melts to acquire a quantified dissolved free oxide concentration, and the zirconium ions were then gradually introduced into the melts in the form of zirconium tetrafluoride (ZrF4) (Sigma-Aldrich, 99.99%) to prepare assorted melts with different nZr/nO. The fluorides in a nickel crucible were placed in a stainless steel cell inside an electric furnace and melted at 600 °C. The temperature was measured by a nickel–chromium thermocouple positioned just outside the crucible. The whole experiment was conducted inside a glove box with dry argon atmosphere (99.99%), and the concentration of moisture and oxygen in the glove box were both under 2 ppm.
| LiF–NaF–KF (46.5:11.5:42 mol%) [FLiNaK] mixture |
Concentration of oxide analyzed by LECO (mol kg−1) |
|---|---|
| Original material | 0.134 |
| After dehydration by HF–H2 | 0.008 |
2.2 Analytical techniques
During experiment, a portion of the melts was withdrawn from the crucible by a nickel sampler (Φ6 mm) for further analysis.The oxide concentration of the extracted samples was determined by carbothermal reduction technique using LECO, according to the work done by Mediaas et al.21,22 A 0.1–0.15 g sample of molten fluorides was sealed in a tin capsule. Then the tin capsule was heated in a graphite crucible to 2700 °C over a short period of time (up to 400 s). The oxides in the sample would completely react with carbon at this temperature to form CO (g) and CO2 (g), which can be detected in an Infrared Radiation (IR) cell. To avoid any systematic error, five samples were chosen from different locations of the melting salts and analyzed respectively. A mean value as a final result was reported.
The zirconium in FLiNaK was determined by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES, Arcos, Spectro Co., Ltd.). The samples were dissolved in the 4% (v/v) HNO3 (Jingrui) and then introduced in the spectrometer.
Raman analysis was performed to identify the structures of salt. The 532 nm line of an argon ion laser with 100 mW average power was used for exciting the sample. For analyzing and collecting the spectra, the HR 800 (Jobin Yvon) Raman system equipped with CCD detector was used in the triple configuration. The system was interfaced with a personal computer and the spectra were saved in digital form. We carried out in situ studies using a Linkam TS1000 environmental cell. The sample was put in a ceramic furnace with a thermocouple in intimate contact. Gas flowing over the sample was regulated by electronic thermal mass flow controllers and the laser was focused on the sample through a water-cooled silica window in the cover of the cell. The in situ studies detailed here used a ramp with rate of 5 °C min−1 to the appropriate temperature, and the temperature was then held to be constant while Raman data were acquired.
3. Results and discussion
3.1 The calibration of LECO
The demonstration of accuracy for LECO was implemented in advance. After addition of different amounts of Li2O into the FLiNaK melts, LECO was used to detect the increments of oxide,23 which should be consistent with the theoretical values. Fig. 1 shows the relation of the theoretical increments vs. detected ones. The slope is approximately equal to 1, which indicates the LECO is reliable to detect the variation of oxide in FLiNaK melts.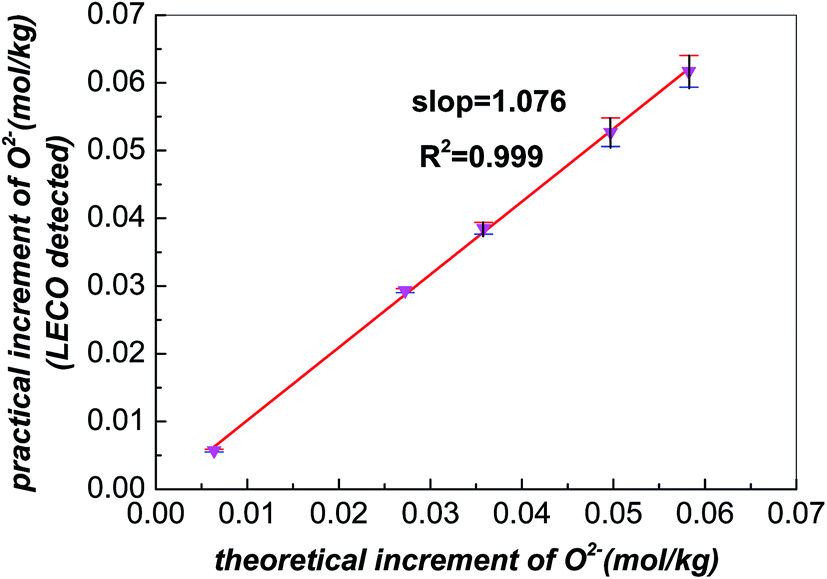 | ||
| Fig. 1 The relation between the theoretical and detected increment of oxide in FLiNaK after addition of different amounts of Li2O. | ||
3.2 Chemical interactions between zirconium and free oxide in molten FLiNaK
| 2O2− + ZrF4 = ZrO2↓ + 4F− | (1) |
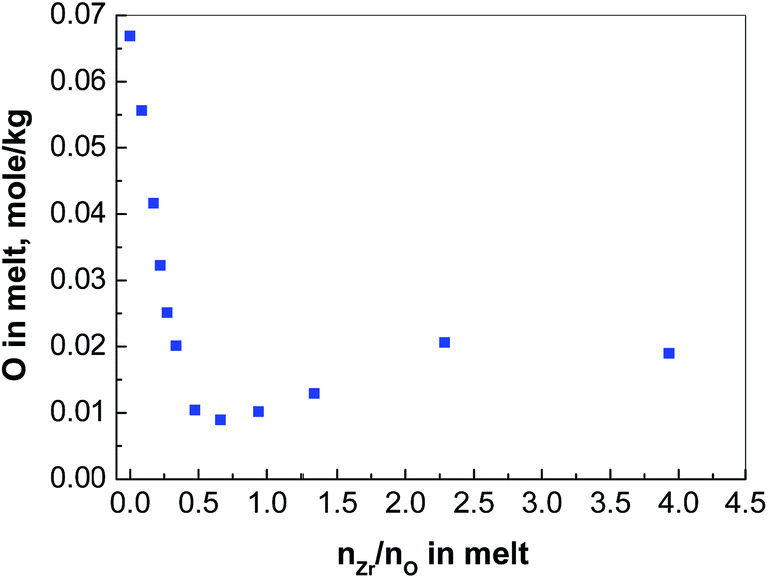 | ||
| Fig. 2 The concentration of oxide in FLiNaK melts versus nZr/nO at 600 °C. | ||
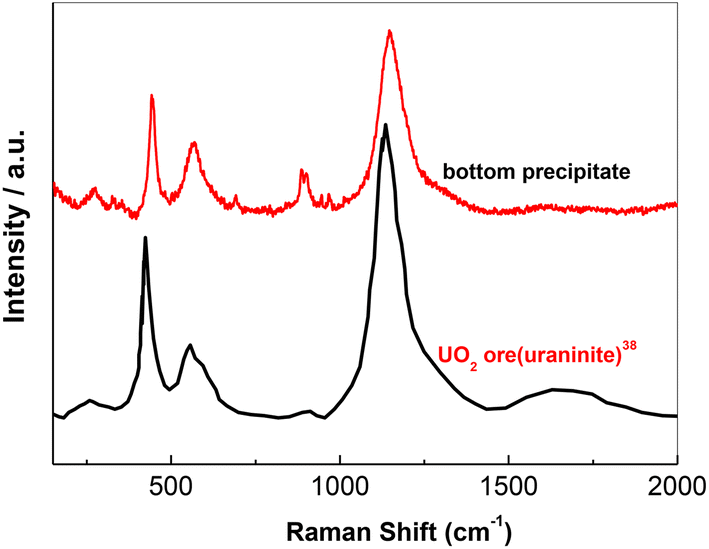
When nZr/nO equals to be 0.5, the whole cylinder block is taken out of the nickel crucible after the molten salt was solidified. A relatively thin layer of precipitates is observed at the bottom (see Fig. 3(a)), which presents a different appearance from that of the rest of the cylinder (Fig. 3(b) shows the upper surface as reference). Then ca. 0.1 g precipitates were sampled and transferred to a sealed high-purity quartz container filled with argon gas for Raman analysis. Fig. 4 shows that the Raman spectrum of the bottom sample is in good agreement with that of the monoclinic ZrO2 (Sigma-Aldrich, 99.99%). Therefore, we can conclude that the precipitates formed with nZr/nO ≤ 0.5 in FLiNaK melts correspond to ZrO2 (see eqn (1)). However, the oxide concentration began to increase when it furthered to add ZrF4, which can be concluded that a solubility equilibrium of ZrO2 may be established. This will be discussed in the next section.
 | ||
| Fig. 3 The solidified FLiNaK melts prepared with initial oxide concentration of 0.067 mol kg−1 and nZr/nO = 0.5; (a) bottom surface; (b) upper surface. | ||
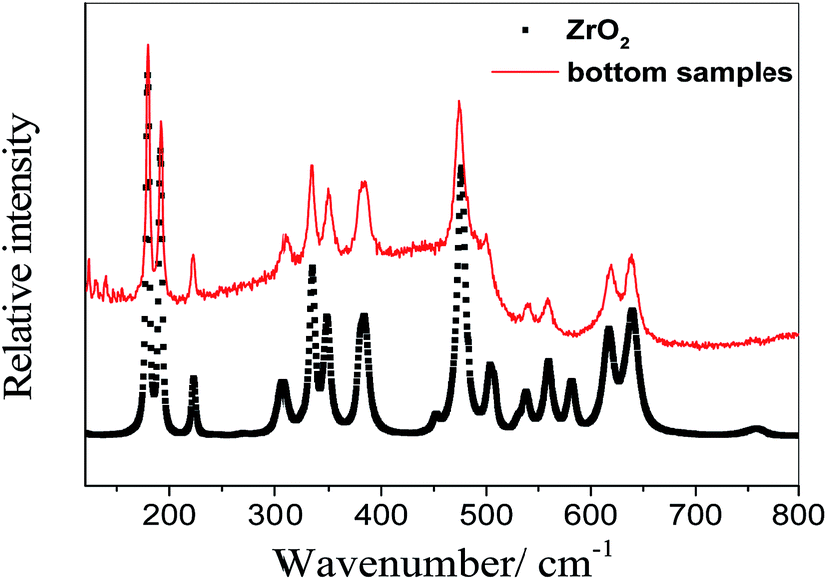 | ||
| Fig. 4 The Raman spectra of monoclinic ZrO2 and the bottom precipitates sampled from the solidified FLiNaK melts at initial oxide concentration of 0.067 mol kg−1 and nZr/nO = 0.5. | ||
After nZr/nO = 3.9 is attained, a nickel rod was quickly inserted into the melts vertically without touching the bottom precipitates and then it was rapidly extracted. During this process, the melts adhered to the rod and experienced a quenching process. Due to its inhomogeneity, two Raman spectra of the sample were measured at 25 °C, with two peaks at 250 and 548 cm−1 in Fig. 5(b) and five peaks at ca. 223, 300, 395, 520 and 548 cm−1 in Fig. 5(d).
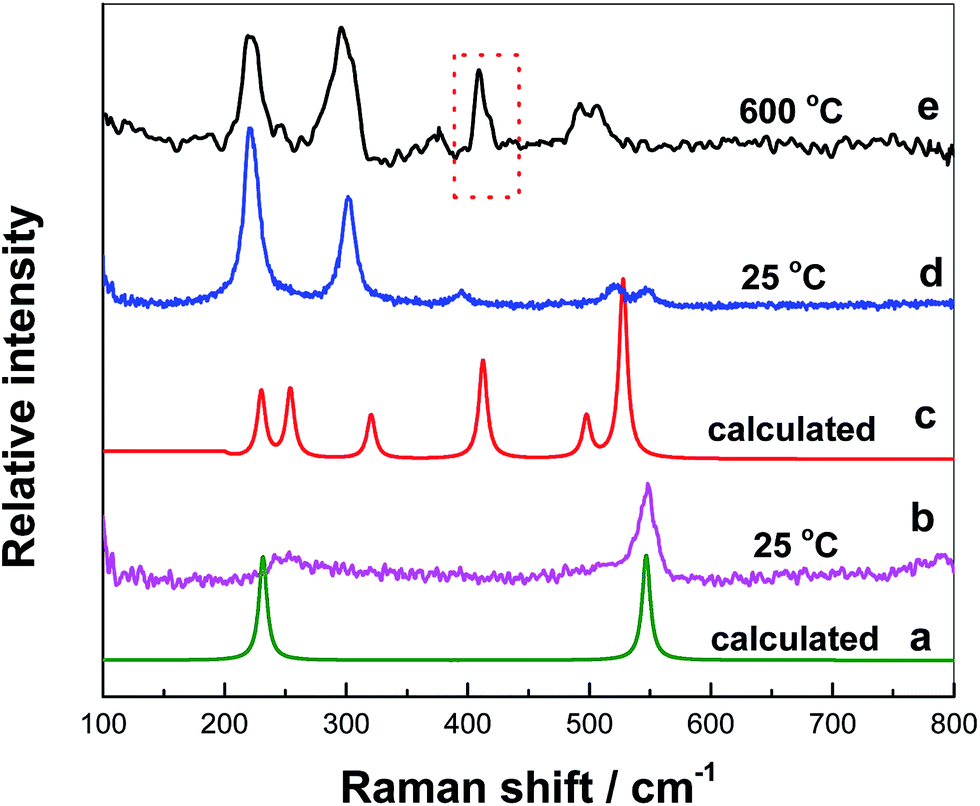 | ||
| Fig. 5 The Raman spectra of (a) the calculated Raman spectrum of ZrF62− complex; (c) the calculated Raman spectrum of Zr2OF104−; (b) and (d) the upper melts extracted from FLiNaK at initial oxide = 0.067 mol kg−1 and nZr/nO = 3.9 measured at 25 °C; (e) measured at 600 °C. | ||
The spectrum of the calculated octahedral [ZrF6]2− structure was shown in Fig. 5(a). It also appeared two peaks at 231 cm−1 and 548 cm−1, corresponding to the bending and symmetrical stretching vibration of Zr–F, respectively, which was consistent with that of Fig. 5(b). Besides, the predominance of octahedral [ZrF6]2− was also detected in KF–ZrF4 melts as reported by Dracopoulos24 and Pauvert.25,26 Thus, the as-detected [ZrF6]2− structure in the melting FLiNaK prepared with nZr/nO = 3.9 is probably formed via the following reaction [eqn (2)].
| 2F− + ZrF4 = ZrF62− | (2) |
The cluster model of F–Zr–O was computed by the quantum chemical ab initio calculations based on the Gaussian 09 software and it was further optimized by the Restricted Hartree–Fock (RHF) method and connection of SDD. The calculated Raman spectrum of [Zr2OF10]4− structure was plotted in Fig. 5(c) and it basically agreed with that of Fig. 5(d). But the peak at 223 cm−1 in Fig. 5(d) was due to the overlapping of the two peaks 230 and 250 cm−1 in Fig. 5(c), corresponding to the F–Zr–O bending vibration and F–Zr–O ω-rocking vibration, respectively. The peaks at 300, 520 and 548 cm−1 were antisymmetric stretching vibration of Zr–F and the peak at 395 cm−1 corresponded to the symmetrical stretching vibration of Zr–F. As the test temperature increased to 600 °C, the sample melted to liquid and became homogenous. The Raman spectrum was shown in Fig. 5(e). The expected trends was observed; i.e. red frequency shifting at high temperature.24 Besides, another peak for F–Zr–O symmetrical stretching vibration was observed at 410 cm−1. This peak was probably attributed to [ZrOF6]4− produced by the decomposition of [Zr2OF10]4− at 600 °C.
The detected [Zr2OF10]4− in the melting FLiNaK was probably formed via eqn (3), and the increment of detected oxide concentration by increasing nZr/nO from 0.7–2.3 (Fig. 2) was also linked to the formation of Zr2OFx6−x complexes.
| ZrO2 + 3ZrF4 + 8F− = 2[Zr2OF10]4− | (3) |
The oxyfluorides in molten fluorides have already been addressed in the field of rare earth27–34 and aluminum production.35,36 Stefanidaki32 found that the solubility of Nd2O3 at 900 °C increased to 0.15, 0.22 and 0.38 mol% when NdF3 in LiF melt was increased to 15, 23 and 30 mol%, respectively. They proved that dissolution of Nd2O3 in NdF3–LiF is attributed to a reaction of Nd2O3 + NdF3 + 3(x − 1)F− = 3NdOFx(x−1)−. Haas37 also suggested that the solubility of UO2 in LiF–BaF2–UF4 or LiF–CaF2–UF4 is strongly influenced by the concentration of UF4 in the melt, which is connected to the possible existence of complex UOxFy. Additionally, the solubility of La2O3 is lower in LiF melts27 than that in LaF3–LiF melts due to the formation of LaOxFy complexes.28,29 In this study, the precipitated ZrO2 also reacted with the excessive ZrF4 and F− at nZr/nO > 0.5 to form Zr2OFx6−x complexes as indicated in eqn (3), which obviously increased the solubility of ZrO2 in the FLiNaK melts. This dissolution mechanism is similar to those reported by Stefanidaki,32 Haas,37 M. Ambrová,27 and Rollet.28,29
3.3 The solubility equilibrium of ZrO2 in FLiNaK–ZrF4 melts
Under the condition of nZr/nO = 3.9, the initial oxide concentration of 0.067 mol kg−1 in FLiNaK melts was finally converted to that of in ZrO2 precipitates and in Zr2OFx6−x complexes. And the Zr2OFx6−x complexes finally stabilized at 0.020 mol kg−1. With an initial oxide concentration of 0.020 mol kg−1 and addition enough amount of ZrF4, one could assume that the free oxide in FLiNaK melts can be completely converted to that in the complexes without precipitating ZrO2. As shown in Fig. 6, the oxide concentration drops sharply from 0.020 to 0.009 mol kg−1 when nZr/nO is increased from 0 to 0.5. With continual addition of ZrF4, the oxide concentration reaches its lowest value 0.009 mol kg−1 and then begins to go up. The curve in Fig. 6 reveals a similar trend as observed in Fig. 2. These observations indicate that the solubility equilibrium of ZrO2 was established.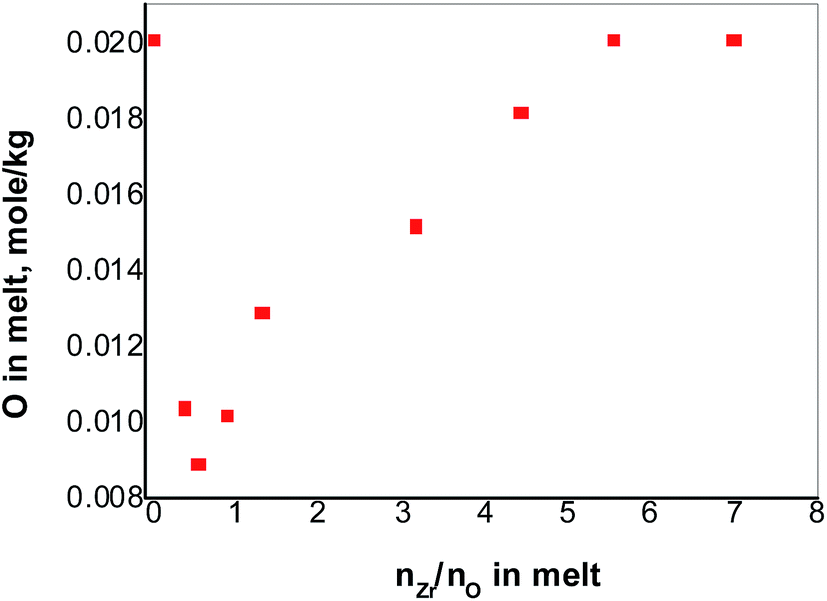 | ||
| Fig. 6 The concentration of oxide in the FLiNaK melts versus nZr/nO at 600 °C. | ||
The descent part of the curve (nZr/nO ≤ 0.5) in Fig. 6 reveals that a reaction occurred between the initial free oxide and the added ZrF4, and ZrO2 precipitates as well as its solubility equilibrium were formed. With the further addition of ZrF4, the oxide concentration reached its minimum value which was 0.009 mol kg−1 and the maximum amount of ZrO2 precipitates was also achieved. Since the solubility equilibrium of ZrO2 was preserved as long as the ZrO2 precipitates existed, it can be concluded that the minimum oxide concentration of 0.009 mol kg−1 was produced by the dissolved ZrO2. According to the stoichiometric relation [eqn (1)], the solubility of ZrO2 in FLiNaK melts equaled to be 0.0045 mol kg−1 (0.019 mol%).
The ascending part of the curve (nZr/nO > 0.5) in Fig. 6 shows that the ZrO2 started to react with the excessive ZrF4 to form Zr2OFx6−x complexes according to eqn (3), and the solubility of ZrO2 in FLiNaK–ZrF4 melts gradually increased as well. Finally, the maximum dissolved oxide of 0.020 mol kg−1 was attained in FLiNaK–ZrF4 melts, indicating that the maximum solubility of ZrO2 was 0.020 mol kg−1 (0.080 mol%), which was much lower than that of Nd2O3 in LiF–NdF3 (ref. 32) and UO2 in CaF2–LiF–UF4 and BaF2–LiF–UF4.37 It is worth mentioning that with an initial oxide concentration less than 0.020 mol kg−1, the free oxide can be fully converted to [Zr2OFx]6−x complexes without any ZrO2 precipitates, given that enough ZrF4 was added.
3.4 The mechanism of ZrF4 protection against the UO2 precipitation
Further validation for the conversion of free oxide to Zr2OFx6−x complexes was carried out. Two sets of FLiNaK melts were prepared by adding Li2O. They both had the same initial concentration of free oxide 0.020 mol kg−1. One set was added with ZrF4 to achieve nZr/nO = 7.0, while the other set was not (nZr/nO = 0). Varied amounts of UF4 (China North Nuclear Fuel Co., Ltd, 99.99%) were then introduced into the two prepared melts, respectively.In the melts prepared with nZr/nO = 0, the oxide concentration detected by LECO was decreased from 0.020 mol kg−1 to 0.016 mol kg−1, while the uranium concentration analyzed by ICP-OES was reduced from 0.007 mol kg−1 to 0.005 mol kg−1 in the first 3 h, as shown in Fig. 7(a). From 3 to 60 h, the decrements of both oxide and uranium gradually diminished and their concentrations reached plateaus at 0.013 mol kg−1 and 0.004 mol kg−1, respectively. Since the decrement ratio of oxide and uranium approximately equals to 2, it can be concluded that the UO2 precipitates were formed from the reaction between UF4 and free oxide, as shown in eqn (4). In addition, a layer of precipitates with rufous color was observed on the bottom surface after solidification, as shown in Fig. 8(a). The Raman spectrum of this precipitates is consistent with that of UO2 reported by Stefaniak et al.,38 as shown in Fig. 9.
| 2O2− + UF4 = UO2↓ + 4F− | (4) |
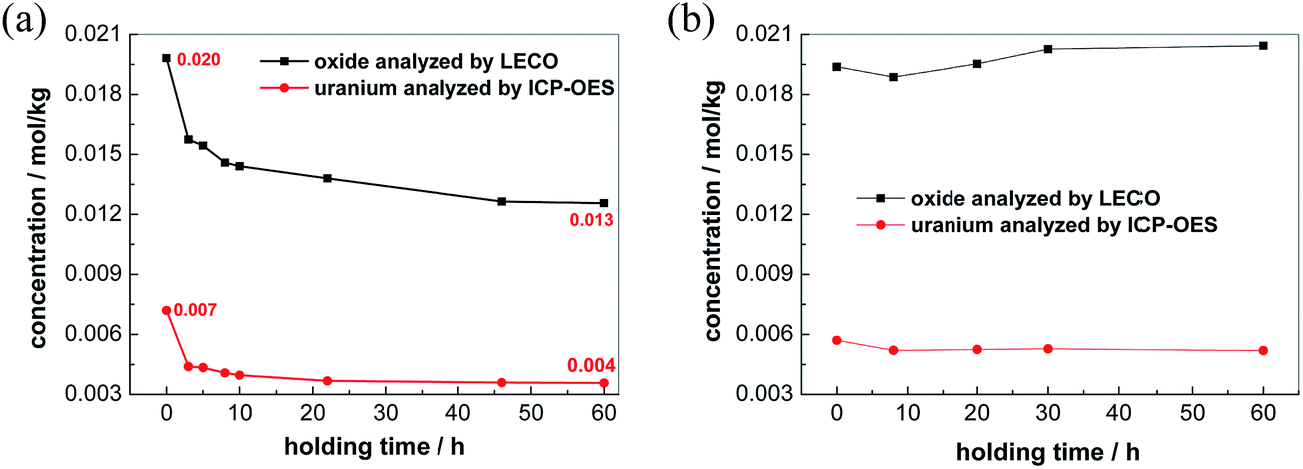 | ||
| Fig. 7 The concentration of oxide and uranium varied with time at 600 °C (a) in FLiNaK melts with initial oxide at 0.020 mol kg−1 and nZr/nO = 0; (b) in FLiNaK melts with initial oxide at 0.020 mol kg−1 and nZr/nO = 7.0. | ||
 | ||
| Fig. 8 The solidified FLiNaK melts with initial oxide at 0.020 mol kg−1 and different nZr/nO; (a) nZr/nO = 0; (b) nZr/nO = 7.0. | ||
 | ||
| Fig. 9 The Raman spectra of UO2 (ref. 38) ore and the bottom precipitates taken from the solidified FLiNaK–UF4 (0.007 mol kg−1) melts at initial oxide = 0.020 mol kg−1 and nZr/nO = 0. | ||
With nZr/nO = 7.0, the concentration of oxide was merely varied in the range of 0.019 mol kg−1 to 0.021 mol kg−1 in 60 h, while the concentration of uranium approximately maintained at 0.005 mol kg−1. Besides, the solidified melt consisted of a small amount of green colored bulk and no precipitates were observed at the bottom surface, as shown in Fig. 8(b). All these indicate that no chemical reaction between oxide and uranium was occurred. Therefore, with an initial oxide concentration less than 0.020 mol kg−1, the free oxide can be fully converted to [Zr2OFx]6−x complexes by introducing enough ZrF4 into FLiNaK melts. The conversion of free oxide to Zr2OFx6−x complexes prevented the formation of UO2 precipitates. This method will provide a very efficient way to control the uranium species against the oxygenation in MSR.
4. Conclusions
This work focused on the interactions between zirconium and free oxide in FLiNaK system. It was found that the initial concentration of oxide which was 0.067 mol kg−1 in FLiNaK melts was finally converted to that of in ZrO2 precipitates at nZr/nO ≤ 0.5 and in Zr2OFx6−x complexes at nZr/nO > 0.5 analyzed by LECO and Raman spectroscopy. The maximum Zr2OFx6−x complexes of FLiNaK melts was found to be 0.020 mol kg−1. When the initial concentration of oxide was less than 0.020 mol kg−1, the free oxide in FLiNaK melts would be completely converted to Zr2OFx6−x complexes without any ZrO2 precipitates, given that enough ZrF4 was added.UF4 was introduced to FLiNaK melts for further validating the conversion of free oxide to Zr2OFx6−x complexes. The results showed that the UO2 precipitates had been formed in the FLiNaK system at nZr/nO = 0, while no chemical reaction between oxide and uranium was occurred at nZr/nO = 7.0. The conversion of free oxide to Zr2OFx6−x complexes prevented the formation of UO2 precipitates. This method will provide a very efficient way to control the uranium species against the oxygenation in MSR.
Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant no. XDA02020400) and the Young Scientists Fund of the National Natural Science Foundation of China (Grant no. 51404237).References
- GIF-002-00, http://gif.inel.gov, December 2002.
- C. W. Forsberg, The Americas Nuclear Energy Symposium (ANES), American Nuclear Society, Miami, Florida, 2002 Search PubMed.
- C. W. Forsberg, P. F. Peterson and L. Ott, Proceedings of the 2004 Americas Nuclear Energy Symposium, Miami Beach, Florida, October 2004 Search PubMed.
- M. Rosenthal, P. Kasten and R. Briggs, Nucl. Appl. Technol., 1970, 8, 107–117 CAS.
- C. Forsberg, Prog. Nucl. Energy, 2005, 47, 32–43 CrossRef CAS PubMed.
- A. Nuttin, D. Heuer, A. Billebaud, R. Brissot, C. Le Brun, E. Liatard, J.-M. Loiseaux, L. Mathieu, O. Meplan and E. Merle-Lucotte, Prog. Nucl. Energy, 2005, 46, 77–99 CrossRef CAS PubMed.
- W. Grimes, Nucl. Technol., 1970, 8, 137–155 CAS.
- L. Toth, U. Gat, G. Del Cul, S. Dai and D. Williams, Review of ORNL's MSR technology and status, Oak Ridge National Lab., TN, United States, Funding organisation: USDOE, Washington, DC (United States), 1996 Search PubMed.
- R. Ross, C. Bamberger and C. Baes, J. Inorg. Nucl. Chem., 1973, 35, 433–449 CrossRef CAS.
- Ed. M. Rosenthal, P. Haubenreich and R. Briggs, ORNL-4812, 1972 Search PubMed.
- J. H. Shaffer, in ORNL-4616, 1971 Search PubMed.
- A. Mathews and C. F. Baes Jr, Inorg. Chem., 1968, 7, 373–382 CrossRef CAS.
- R. B. Briggs, in ORNL-3122, 1960 Search PubMed.
- E. G. B. W. R. Grimes, H. F. McDuffie, G. M. Watson, F. F. Blankenship and C. H. Secoy, in ORNL-4076, 1966 Search PubMed.
- J. Shaffer, W. Grimes and G. Watson, J. Phys. Chem., 1959, 63, 1999–2002 CrossRef CAS.
- P. E. Field and J. H. Shaffer, J. Phys. Chem., 1967, 71, 3218–3222 CrossRef CAS.
- D. A. Petti, G. Smolik, M. F. Simpson, J. P. Sharpe, R. Anderl, S. Fukada, Y. Hatano, M. Hara, Y. Oya and T. Terai, Fusion Eng. Des., 2006, 81, 1439–1449 CrossRef CAS PubMed.
- P. Calderoni, P. Sharpe, H. Nishimura and T. Terai, J. Nucl. Mater., 2009, 386, 1102–1106 CrossRef PubMed.
- M. Korenko, M. Straka, J. Uhlíř, L. Szatmáry, M. Ambrová and M. Šimurda, J. Radioanal. Nucl. Chem., 2014, 302, 549–554 CrossRef CAS.
- M. Gibilaro, L. Massot, P. Chamelot, L. Cassayre and P. Taxil, Electrochim. Acta, 2013, 95, 185–191 CrossRef CAS PubMed.
- H. Mediaas, J. Vinstad and T. Østvold, Solubility of MgO in MgCl {sub 2}–NaCl–NaF melts, Minerals, Metals and Materials Society, Warrendale, PA, United States, 1996 Search PubMed.
- G. Kipouros, H. Mediaas, J. Vindstad, T. Oestvold and O. Tkatcheva, Oxide solubilities and phase relations in the system Mg–Nd–O–Cl, Minerals, Metals and Materials Society, Warrendale, PA, United States, 1996 Search PubMed.
- P. Chamelot, P. Palau, L. Massot, A. Savall and P. Taxil, Electrochim. Acta, 2002, 47, 3423–3429 CrossRef CAS.
- V. Dracopoulos, J. Vagelatos and G. Papatheodorou, J. Chem. Soc., Dalton Trans., 2001, 1117–1122 RSC.
- O. Pauvert, D. Zanghi, M. Salanne, C. Simon, A. Rakhmatullin, H. Matsuura, Y. Okamoto, F. Vivet and C. Bessada, J. Phys. Chem. B, 2010, 114, 6472–6479 CrossRef CAS PubMed.
- O. Pauvert, M. Salanne, D. Zanghi, C. Simon, S. Reguer, D. Thiaudière, Y. Okamoto, H. Matsuura and C. Bessada, J. Phys. Chem. B, 2011, 115, 9160–9167 CrossRef CAS PubMed.
- M. Ambrová, J. Jurišová, V. Danielik and J. Gabčová, J. Therm. Anal. Calorim., 2008, 91, 569–573 CrossRef.
- A. L. Rollet, H. Matsuura and C. Bessada, Dalton Trans., 2015, 522–529 RSC.
- A. L. Rollet, E. Veron and C. Bessada, J. Nucl. Mater., 2012, 429, 40–47 CrossRef CAS PubMed.
- M. Gibilaro, L. Cassayre, O. Lemoine, L. Massot, O. Dugne, R. Malmbeck and P. Chamelot, J. Nucl. Mater., 2011, 414, 169–173 CrossRef CAS PubMed.
- M. Gibilaro, J. Pivato, L. Cassayre, L. Massot, P. Chamelot and P. Taxil, Electrochim. Acta, 2011, 56, 5410–5415 CrossRef CAS PubMed.
- E. Stefanidaki, G. M. Photiadis, C. G. Kontoyannis, A. F. Vik and T. Østvold, J. Chem. Soc., Dalton Trans., 2002, 2302–2307 RSC.
- A. F. Vik, V. Dracopoulos, G. N. Papatheodorou and T. Østvold, J. Alloys Compd., 2001, 321, 284–299 CrossRef CAS.
- J. Von Barner, E. Christensen, N. Bjerrum and B. Gilbert, Inorg. Chem., 1991, 30, 561–566 CrossRef CAS.
- V. Lacassagne, C. Bessada, P. Florian, S. Bouvet, B. Ollivier, J.-P. Coutures and D. Massiot, J. Phys. Chem. B, 2002, 106, 1862–1868 CrossRef CAS.
- C. Malherbe, G. Eppe and B. Gilbert, Anal. Chem., 2014, 86, 8073–8081 CrossRef CAS PubMed.
- P. A. Haas and S. P. Cooper, J. Chem. Eng. Data, 1993, 38, 26–30 CrossRef CAS.
- E. A. Stefaniak, A. Alsecz, I. E. Sajó, A. Worobiec, Z. Máthé, S. Török and R. Van Grieken, J. Nucl. Mater., 2008, 381, 278–283 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |