
Pd/mannose promoted tandem cross coupling-nitro reduction: expedient synthesis of aminobiphenyls and aminostilbenes†
Abstract
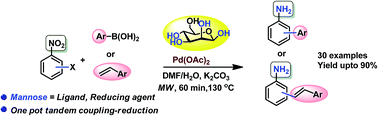
The dual role of D-mannose as a ligand for Pd catalyzed cross-coupling, and as a hydrogen source for nitro reduction is demonstrated in a modular cross coupling-nitro reduction sequence. The synthetic utility and generality of this green protocol has been illustrated by the synthesis of 20 aminobiphenyl and 10 aminostilbene derivatives in high yields through a one-pot Suzuki coupling-nitro reduction and a Heck coupling-nitro reduction, respectively, starting from halonitroarenes as substrates.
 Please wait while we load your content...
Please wait while we load your content...

