
A microfluidic approach to study the effect of bacterial interactions on antimicrobial susceptibility in polymicrobial cultures†
Ritika Mohan,
Chotitath Sanpitakseree,
Amit V. Desai,
Selami E. Sevgen,
Charles M. Schroeder and
Paul J. A. Kenis*
Department of Chemical & Biomolecular Engineering, University of Illinois, Urbana-Champaign, Urbana, USA 61801. E-mail: kenis@illinois.edu; Tel: +1 217 265 0523
First published on 9th April 2015
Abstract
Polymicrobial infections are caused by more than one pathogen. They require antimicrobial dosing regimens that are different from those prescribed for monomicrobial infections because these interactions are predicted to influence the antimicrobial susceptibility of the individual pathogens. Here we report on a microfluidic approach to study the effect of bacterial interactions in polymicrobial cultures on the antimicrobial susceptibility. The use of microfluidics enables real-time quantification of bacterial growth dynamics in the presence and absence of antimicrobials, which is challenging to achieve using current methods. We studied microbial interactions between Pseudomonas aeruginosa, and Escherichia coli and Klebsiella pneumoniae. A key observation was that in co-cultures with relatively high initial cell numbers of P. aeruginosa, the co-cultured partner bacteria exhibited initial growth followed by lyses or growth stasis. In addition, we observed a significantly higher antimicrobial tolerance of P. aeruginosa in polymicrobial cultures, as evident by up to 8-fold increases in the minimum inhibitory concentration of the antimicrobials, compared to those observed in monomicrobial cultures. This work demonstrates the potential of microfluidics to study bacterial interactions and their effect on antimicrobial susceptibility, which in turn will aid in determining appropriate antimicrobial treatment for polymicrobial infections.
Introduction
Microbes seldom exist in isolation.1,2 Several bacterial infections that affect humans such as urinary tract infections (UTIs), chronic wounds, cystic fibrosis, and nosocomial bacteremia are polymicrobial infections.3–7 The pathogenesis and virulence of many more infections are speculated to be influenced (generally enhanced) by interactions among different bacterial species.6,8–10 These infections, also referred to as mixed, complex, synergistic, or co-infections, are known for their higher mortality rate compared to monomicrobial infections involving only one of the bacterial species.4 Despite their larger debilitating effects, many polymicrobial infections are still not well understood.4,5,7,11Polymicrobial infections frequently require stronger antimicrobial dosing regimens because microbes involved in polymicrobial infections are more recalcitrant to antimicrobials compared to the same microbes in monomicrobial cultures.10 The majority of the treatments for polymicrobial infections, however, are solely based on monomicrobial susceptibility information,12–14 despite the fact that monomicrobial antimicrobial susceptibility information does not generally apply to polymicrobial cultures.15–18 The prime reason for this lack of translation is that the interactions among different bacteria species influence the response of the bacterial to antimicrobials.6,8,10 Hence, incorrectly assumed correlations between the susceptibility of mono- and polymicrobial infections may lead to inadequate antimicrobial dosing regimens, which also further exacerbates the global issue of increasing antimicrobial resistance.19 The first step towards addressing these issues of inadequate dosing and rising resistance is improving our understanding of the influence of inter-species microbial interaction on the antimicrobial susceptibility of the individual microbes involved in these polymicrobial infections, which would lead to more effective treatment.6 Moreover, research on bacterial interactions will aid the discovery of signalling molecules that are produced by individual pathogens in the presence of other species, which may modulate the physiology and possibly the pathogenesis of those other microbes.14,20–23
Conventional antimicrobial susceptibility testing (AST) procedures such as disk diffusion and broth dilution typically provide useful insight about bacterial susceptibility in isolate cultures,24 but they are not well suited to obtain analogous, species-specific antimicrobial susceptibility information in mixed cultures. When employed as end-point assays, these methods do not provide quantitative information on the dynamics of bacterial interactions during growth, and they do not allow for observation of inter-species interactions, and their influence on the susceptibility of the individual microbes.
Despite these limitations, a few attempts to adapt conventional AST methods for polymicrobial cultures have been reported. A recent study involving P. aeruginosa, B. cenocepacia, and E. coli demonstrated an increase in the antimicrobial resistance of the co-cultured population compared to the monomicrobial cultures, which was attributed to the transfer of resistance from a resistant sub-bacterial population.25 Because the bacterial interactions can influence susceptibility, a few of these studies have focused on the effects of bacterial interaction on antimicrobial susceptibility in polymicrobial cultures.26,27 In these studies, changes in antimicrobial susceptibility of polymicrobial cultures (B. fragilis, E. coli and E. faecalis) have been primarily attributed to the production of enzymes such as β-lactamases, which protect co-existing bacteria. A few studies also have focused on manipulation of inter-species interaction to influence susceptibility. Addition of clavulanic acid, which is known to potentiate the activity of amoxicillin and ticarcillin, was shown to decrease the susceptibility of the β-lactamase-producing bacteroides species in co-cultures.28 Interaction between different microbes can also be manipulated by targeted mutation of the genes responsible for inter-species communication. Studies have shown that suppression of this communication, induced by suppression of quorum sensing and sensing of other bacterial species, leads to a decrease in the resistance to antimicrobials in mixed cultures of P. aeruginosa and E. coli.8,10
While these previous studies provided some insights into polymicrobial infections, they are still hampered by some of the shortcoming of the conventional, culture-dependent methods, including long analysis times (>24 h), low detection sensitivity,29,30 and the inability to determine species-specific susceptibility. To overcome some of these issues, culture-independent methods, where the bacteria is not required to be pre-cultured prior to the study, have been used for polymicrobial culture studies.6 One such method is the quantitative terminal restriction fragment length polymorphism (qT-RFLP) approach that enumerates the 16S rRNA gene as a proxy for bacterial cell number. In mixed cultures comprising P. aeruginosa, B. cepacia, and S. aureus, qT-RFLP was used to quantify the time required for stable-coexistence (a study not possible with culture-dependent techniques),31 and to study the effect of inter-species interaction on antimicrobial resistance to ceftazidime.24,32
The use of integrated microfluidic platforms is another promising, culture-independent method for the study of microbial infections. In fact microfluidics has been used previously to study a variety of phenomena in bacterial populations such as the influence of physical (shear stresses) and chemical (toxins) cues on cell viability, motility, functionality, inter-species interaction, and proliferation.14,33–38 Over the last six years, microfluidic approaches have been used for monomicrobial AST studies, exploiting the much smaller sample volumes (<1 mL) needed and the ability to test many different conditions in a combinatorial fashion.39–46 More importantly, microfluidic approaches allow for detection of bacterial cells with high sensitivity, which obviates the need for pre-culturing of the bacteria for long periods, thus drastically reducing the assay time. For example, a droplet-based microfluidic approach was used to determine the minimum inhibitory concentrations (MIC) of ampicillin, tetracycline, and chloramphenicol (pair-wise and individually) against E. coli.41 To address some of the limitations of droplet-based microfluidic platforms (e.g., the instability in droplet formation), microfluidic platforms with continuous flow have been developed. For instance, a high-throughput microfluidic platform comprising 32 micro-wells was used to determine the antimicrobial susceptibility of E. coli to tetracycline and erythromycin.45
Yet others developed a portable microfluidic platform, potentially suitable for point-of-care (POC) application, able to determine the MICs of vancomycin, tetracycline, and kanamycin against E. faecalis, P. mirabilis, K. pneumoniae, and E. coli against.42 In a related approach, we reported application of a microfluidic array platform to determine MICs of four antibiotics (ampicillin, cefalexin, chloramphenicol, and tetracycline) and their combinations against E. coli.46 This platform uses only ∼2.4 nL per condition tested and can provide MICs in less than 4 hours in certain cases, much shorter than the 1–3 days needed with some of the methods currently used for similar research. The latter microfluidic approach also addresses several limitation of the prior microfluidic approaches for monomicrobial AST, including limited portability41,44,47 or complicated platform fabrication and/or operation.44,48
Here we report the application of a multiplexed microfluidic platform to study polymicrobial cultures. In addition to benefiting from the aforementioned advantages of microfluidics for monomicrobial AST studies, polymicrobial AST studies benefit from the use of microfluidic platforms due to the ability to quantify bacterial interactions and ability to determine species-specific susceptibility by distinguishing and accurately enumerating different species in a time-resolved fashion. As a result, the approach allows for accurate determination of bacterial growth curves of individual species. Being able to acquire accurate time–kill curves (i.e., precise specifies-specific antimicrobial susceptibility information) is important for determining effective treatment of bacterial infections.49 Here we demonstrate the utility of the microfluidic approach by studying the interactions between E. coli, P. aeruginosa, and K. pneumoniae in the absence and presence of commonly used antimicrobials.
Experimental
Fabrication of the microfluidic platform
We used standard two-layer soft lithography to fabricate the microfluidic devices (Fig. S1†).50 The device consists of a thick control layer of poly(dimethylsiloxane) or PDMS (∼10 mm) bonded to a thin fluidic layer of PDMS (∼35 μm). Briefly, photolithography was used to pattern the control and fluid layer designs on silicon wafers using negative photoresist SU-8 25 purchased from MicroChem Corporation (Newton, MA) to create master patterns. A thin layer of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (weight of monomer to cross linker) PDMS was spin coated on fluid layer master (∼35 μm). Separately, a thick layer of 5:1 PDMS was poured over the control layer master. The fluid and control layers were then baked at 65 °C for 30 minutes to cure the PDMS. Next, the control layer was peeled off from the master, and control line holes were punched using a 20-gauge needle. The control layer was then manually aligned to the fluid layer master under a microscope (Leica MZ6) to assemble an aligned two-layer device. This aligned device was then cured at 65 °C overnight and carefully peeled off the fluid layer master. Finally, inlet holes were punched in the device using a 20-gauge needle, and the integrated two-layer device was placed on a cleaned glass coverslip (no. 1.5) purchased from Ted Pella, Inc. The procedure for cleaning the coverslips is detailed in the ESI.†
:1 (weight of monomer to cross linker) PDMS was spin coated on fluid layer master (∼35 μm). Separately, a thick layer of 5:1 PDMS was poured over the control layer master. The fluid and control layers were then baked at 65 °C for 30 minutes to cure the PDMS. Next, the control layer was peeled off from the master, and control line holes were punched using a 20-gauge needle. The control layer was then manually aligned to the fluid layer master under a microscope (Leica MZ6) to assemble an aligned two-layer device. This aligned device was then cured at 65 °C overnight and carefully peeled off the fluid layer master. Finally, inlet holes were punched in the device using a 20-gauge needle, and the integrated two-layer device was placed on a cleaned glass coverslip (no. 1.5) purchased from Ted Pella, Inc. The procedure for cleaning the coverslips is detailed in the ESI.†
Bacterial strains, media and antimicrobials
Bacterial strains of E. coli, P. aeruginosa and K. pneumoniae were routinely cultivated in Lennox broth (10 g L−1 tryptone, 5 g L−1 yeast extract, and 5 g L−1 NaCl) supplemented with antimicrobials for selective growth, as shown in Table 1. Wild type E. coli MG1655 cells were transformed with a plasmid pAM06 (ref. 51) (conferring kanamycin resistance) to constitutively express the green fluorescent protein (GFP) under control of the PL promoter from phage lambda. K. pneumoniae strain 342 carrying plasmid pRK2073 (ref. 52) (conferring kanamycin resistance) constitutively expressing GFP was generously provided to us by Prof. Eric Triplett at the University of Florida. P. aeruginosa expressing a red fluorescent protein (RFP) with the tdtomato gene under the transcriptional control of the nptll promoter on pBBR1-based plasmid pMQ132 (ref. 53) (conferring gentamicin resistance) was generously provided by Prof. Robert Shanks at the University of Pittsburgh. Incorporation of GFP and RFP markers enables facile detection and counting of bacterial cells using time-lapse fluorescence microscopy. Prior to all monomicrobial and polymicrobial experiments, frozen bacterial stocks were revived overnight on LB agar plates supplemented with appropriate antimicrobials. Single colonies from plates were picked and used to inoculate 5 mL LB broth cultures, which were incubated overnight at 37 °C with aeration (200 rpm).| Pathogen | Supplemented antimicrobial |
|---|---|
| pQE80L-PLGFP/E. coli | 30 μg mL−1 kanamycin |
| pRK2073-GFP/K. pneumoniae | 30 μg mL−1 kanamycin |
| pMQ132-Pnptlltdtomato/P. aeruginosa | 40 μg mL−1 gentamicin |
| Wild-type E. coli | No antimicrobial |
For preparation of polymicrobial cultures (Fig. S2†), 50 μL of the two monomicrobial cultures (cell types 1 and 2) were inoculated into separate tubes containing 5 mL of LB without supplemental antimicrobials and were incubated for three hours. Incubated cultures were then concentrated 10× by centrifugation (3200 × g for 10 min) followed by re-suspension in 500 μL of LB, which removed antimicrobials from the cultures.
A range of cell type 1 and type 2 concentrations was then prepared by diluting in LB as shown in Fig. S2.† Finally, diluted cultures of the two types were then mixed to obtain a range of polymicrobial combinations. In the preparation of polymicrobial cultures, we ensured that the observations of the competition experiments were solely due to interactions between bacterial cells rather than antimicrobial–cell interactions, because antimicrobials were removed by centrifugation and by utilizing LB without antimicrobials for subsequent dilution steps. In our experiments, we did not observe a change in cellular fluorescence after cells were sub-cultured in LB without antimicrobials.
Antimicrobial stock solutions of 10 mg mL−1 gentamicin sulfate salt, 10 mg mL−1 tobramycin, 30 mg mL−1 kanamycin sulphate, and 10 mg mL−1 amikacin were prepared in sterile deionized water. Antimicrobial stock solutions were filtered using a 0.45 μm syringe filter (Millex-HV filter unit, Millipore) prior to use. Gentamicin sulfate and tobramycin were purchased from Sigma-Aldrich, kanamycin sulfate was purchased from Invitrogen, and amikacin was purchased from Fisher Scientific.
Off-chip AST
To compare the MIC obtained on-chip to conventional methods, we performed AST against E. coli, K. pneumoniae, and P. aeruginosa using 96-micro-well plates, the volume of each well was 360 μL, using the broth dilution method or the conventional two-fold dilution protocol. The monomicrobial cell cultures were prepared as described previously46 and polymicrobial cultures were prepared as shown in Fig. S2.† Bacterial solution (100 μL) and antimicrobial solution (100 μL) were added to the wells of a 127.8 mm × 85.5 mm flat-bottom 96-well plate (Nunclon), and the plates were incubated overnight at 37 °C. MICs were determined by visual inspection of the bacterial density (i.e., observed for cloudiness) in each well, where the minimum concentration of antimicrobial that prevented observable cell growth (no observable cloudiness) was defined as the MIC. Antimicrobial concentrations were prepared by the two-fold dilution protocol, so the precision of the method is considered to be plus or minus one two-fold concentration.49 In some cases, we report a range of MIC values because occasionally a particular antimicrobial concentration would allow for only marginal cell growth, whereas the next higher concentration would cause cell death. Note that sometimes in the broth dilution method, the cloudiness is determined using microplate readers, where the optical absorbance values are used as an indirect estimate of the cell numbers (optical density or OD measurements). Although this procedure of using OD measurement is more accurate than the one using visual inspection, absorbance values are still an indirect estimate of the number of cells and hence the broth dilution method is less quantitative than the microfluidic approach which provides the ability to track and enumerate individual bacterial cells.On-chip AST
Microfluidic devices were sterilized by autoclaving prior to each experiment, and the flow channels were treated with 10 mg mL−1 bovine serum albumin (BSA) for 15 minutes to prevent nonspecific interactions between the surface and cells. The experimental set up and operation of the platform is an adaptation of previously described protocols with the exception of using a 20× objective (Plan Achromat, NA = 0.40) for image acquisition, which allows for better resolution of cells at higher cell densities compared to 10× objective.46 The imaging and data acquisition process is completely automated and the details have been reported previously.46 Briefly, ImagePro Plus software was used to move a programmable stage to every well in the platform to allow for acquisition of fluorescent images (with 20× objective) for every time point. A limitation of the automated focusing routine to identify the plane of maximum focus is that the cells outside the DOF will not be imaged and thus not included in the enumeration. By manually traversing the z-direction we observed, however, that the majority of the cells were present within a single plane of focus and hence the cell population in this plane is a sufficiently accurate representation of the total cell population.Images obtained using fluorescence microscopy were analyzed (Fig. S3†) and post processed using ImageJ (Version 1.47c). The number of cells in each chamber was determined using local fluorescence intensity maxima, as previously described.46 Fig. S4 and S5† depict fluorescent images of the co-cultures for different conditions. Briefly, we used the intrinsic ImageJ function ‘Enhance Contrast’ to enhance image contrast followed by the ‘Find Maxima’ function was used to determine the number of cells by counting the local fluorescence intensity maxima. This method works well as long as most cells can be easily identified, as we confirmed by comparing manual counts with the automatically obtained counts for several images. Enumerating cell populations with very high cell densities will not be accurate due to overlapping cells in the z-direction. Finally, we plotted time–kill curves of the bacteria for different antimicrobials (cell number versus time). To better highlight long-term trends in cell growth (or death); we implemented a two-point moving average filter, which tends to smooth the time–kill curves. In case of on-chip experiments, we define the MIC as the concentration value of the antimicrobial where the number of cells after 16 hours is less than the initial number of cells. This definition ensures consistency with MICs obtained using off-chip experiments, where the MIC is determined by visually inspecting the 96-well plate after an overnight culture (∼16 hours). Fig. S6 and S7† depict examples of the time–kill curves. Note that in certain cases we observe cell growth initially followed by cell lyses such that the final cell numbers after 16 hours is less than the initial cell number; this concentration value of the antimicrobial will still be defined to be the MIC. In Table 2, we report the comparison between MIC values for tobramycin obtained using off-chip and on-chip experiments.
| Amikacin [μg mL−1] | Tobramycin [μg mL−1] | |||
|---|---|---|---|---|
| Bulk | On-chip | Off-chip | On-chip | |
| E. coli | 8 | 4 | 8 | 4 |
| K. pneumoniae | 8 | 8–16 | 4–8 | 4–8 |
| P. aeruginosa | 2–4 | 8 | 2 | 4–8 |
Results and discussion
Design and validation of the platform
In this work we use a two-layer PDMS microfluidic platform to study the interaction between different bacterial species in the presence and absence of antimicrobials (Fig. 1). The microfluidic assembly (an improved version of a platform we have used previously for monomicrobial AST studies46) consists of: (1) a control layer for actuating the mixing and filling valves, and (2) a fluidic layer that contains the flow channels and 48 wells (4.8 nL each). Each well comprises two square half-wells that are 400 μm by 400 μm in dimension. Individual wells can be fully visualized within the field of view of an inverted microscope using an objective with 20× magnification. Bacterial cells could be readily visualized in the transverse direction (z-direction) using an automated focusing routine, because the flow channel height was optimally designed to be 15 μm, which is sufficiently high to not physically stress the bacterial cells and sufficiently low to constrain the majority of the cells within a single focal plane.46 In particular, the depth of field (DOF) for imaging was determined to be ∼6 to 8 μm, and the experiments were performed by using the automated focusing center, which typically identified the center plane of the fluidic channel as the plane of maximum focus. In this way, the majority of the cells remained within the DOF in the z-direction during data acquisition, which we also confirmed by manually traversing the z-direction for some cases. This capability provided by the microfluidic approach to track and enumerate cells in micro-wells with high resolution obviates the need to pre-culture the bacteria to the large numbers typically required using conventional methods (∼107 cells), thus drastically reducing the assay time by eliminating the lengthy pre-culturing step (1–3 days). For polymicrobial susceptibility testing, each half-well contained bacterial cells of one or more bacterial species, and the adjacent half-well contained the antimicrobial solutions. This arrangement allows for a set of 12 unique conditions to be tested in quadruplicates (48 experiments) on a single microfluidic platform. | ||
| Fig. 1 Optical micrographs of the microfluidic platform for quantifying on-chip polymicrobial interactions and the susceptibility of bacteria to antimicrobials in polymicrobial cultures. (A) 48 well-array chip wherein each well is comprised of two 2.4 nL half-wells, here filled with dyed aqueous solutions. (B) Close up 48-well array able to screen 12 unique conditions in quadruplicates. For example: 12 unique antimicrobial concentrations and/or different antimicrobial combinations can be loaded in each well through fluid lines (green solutions) and 12 unique polymicrobial bacterial cell solutions can be loaded in the adjacent wells (red solutions). (C) Antimicrobial and bacterial cell chambers are isolated by mixing valves. (D) Same set of wells after opening of the mixing valves, which results in uniform diffusional mixing of the antimicrobials and cells in adjacent chambers, here represented by the dark red solution. | ||
For bacterial interaction studies, each half-well contained a unique combination of bacterial species (in terms of cell number and/or cell type), so that 24 unique conditions can be tested in quadruplicates (96 experiments) on a single platform. Each half-well is isolated from the remaining wells by normally-closed valves, which enhances the portability of the platform by circumventing the need for continuous actuation during the experiment.54 The current platform design is an improvement over our prior platform,46 such that the position of the quadruplicate half-wells are equidistant from their respective inlets. This configuration ensures uniform distribution of cell numbers in each of the quadruplicate half-wells due to the equal hydraulic resistance between the inlet and each of the half-well (Fig. 1). Furthermore, the microfluidic platform reported here enables testing of 96 conditions simultaneously, and all the wells can be imaged in less than 5 minutes for each time point. To further enhance throughput, more wells can be added (i.e., to test more conditions) to the platform. Although integration of additional wells will lead to more complex designs and will make the operation a bit more involved, precedents for densely integrated microfluidic array chips exist in the literature.55
In a first set of experiments, we validated the multiplexed microfluidic platform by comparing the MIC of amikacin and tobramycin (Table 2) obtained in on-chip AST experiments to those obtained using a conventional AST method (micro-broth dilution), against E. coli, P. aeruginosa, and K. pneumoniae individually (monomicrobial AST). The latter two bacteria along with E. faecium, S. aureus, A. baumanii, and Enterobacter species belong to the ESKAPE category, widely known to “escape” the bactericidal action of certain common antimicrobials due to acquired resistance to several antimicrobials.43 The MIC values obtained using micro-broth dilution (off-chip) and on-chip experiments were found to be in close agreement (Table 2). The discrepancies in some cases (off by 2-fold) between the off-chip and on-chip outcomes can be attributed to the inherent differences in the analysis procedures. In on-chip experiments, the MICs are determined by counting the actual changes in cell numbers, whereas in off-chip experiments (using the procedure reported in the Experimental section) the MICs are determined by visually detecting changes in bacterial growth (based on cloudiness of the microwell). The advantages of the on-chip approach over the off-chip method include rapid determination of MIC (2–4 h) using small sample volumes (<3 nL), real-time monitoring of growth dynamics, high sensitivity, high-throughput, amenability to automation, and ease of operation. These on-chip monomicrobial AST results using amikacin and tobramycin against three different species validate that the 48-well microfluidic platform used in this study is suitable for determining accurate MICs.
Dynamics of interaction between P. aeruginosa and E. coli (absence of antimicrobials)
Next we utilized the microfluidic platform to quantify bacterial interaction in polymicrobial cultures over time. P. aeruginosa and E. coli are ubiquitous and often co-exist in many cases of polymicrobial bacterial infections.8 We employed the microfluidic platform to monitor changes in cell numbers using time-lapse fluorescence microscopy (TLFM) in mixed cultures of P. aeruginosa and E. coli expressing RFP and GFP, respectively. The use of GFP and RFP as genetically encodable indicators of cell viability has been reported previously.56,57 We prepared co-cultures of P. aeruginosa and E. coli prior to introducing them in the microfluidic platform (Fig. S2†). Co-cultures were prepared such that the initial cell numbers of P. aeruginosa and E. coli varied across a wide range (10 to 900) to study diverse cases of bacterial interactions that can occur in mixed infections. Pre-mixed cells were introduced in the microfluidic platform, and TLFM was used to visualize and quantify cell numbers of different bacterial species over a period of 16 h (Fig. 2). | ||
| Fig. 2 On-chip real-time monitoring of the interaction between P. aeruginosa (P) and E. coli (E) in the absence of antimicrobials. The plot represents growth curves for (A) P. aeruginosa and (B) E. coli for different starting cell numbers. Cell growth and death were monitored by counting cells in each well, every 30 minutes, over a period of 16 h. For the control curve (isolate), the initial cell numbers for P. aeruginosa and E. coli were 280 and 90. The curves are generated from data points that represent the mean of the measurements from three experiments; but we depict only the guide-lines between the data points for purposes of clarity. (C) Time-lapse fluorescent images for the case of co-culture comprising 90 P. aeruginosa cells (expressing red fluorescent protein or RFP) and 190 E. coli cells (expressing green fluorescent protein or GFP) initially (0 h). | ||
At higher initial cell numbers of P. aeruginosa relative to E. coli (220 and 60), we observed that E. coli is completely eradicated within 8 hours (Fig. 2(B)). P. aeruginosa is hypothesized to produce high concentrations of toxic metabolites such as pyocyanin, which are known to have antimicrobial properties against E. coli.58 We tested this hypothesis by mixing supernatant of an overnight P. aeruginosa culture with E. coli, and observed drastic reduction in growth of E. coli suggesting the toxic effects of the metabolites generated by P. aeruginosa on the cell cultures of E. coli (data not shown). However, in a co-culture of P. aeruginosa and E. coli mixed initially at a 1:1 ratio of cell numbers we observe an initial growth of E. coli followed by growth arrest in 7–8 hours, irrespective of starting the experiment with high (∼800) or low (∼10) total cell numbers of both species. Similarly, at higher initial cell numbers of E. coli (∼190 and 850) compared to those of P. aeruginosa (∼90 and 30), the growth of E. coli levels off after 6 to 8 hours. A possible reason for this behavior could be the production of indole by E. coli.8 Indole enables E. coli to grow in mixed populations potentially including P. aeruginosa by inhibiting pyocyanin production and consequently disrupting quorum sensing in P. aeruginosa.8 In gram negative bacteria such as E. coli, cell-to-cell communication based on cell density is referred to as quorum sensing, which occurs through the release of fatty-acid-based molecules, known as autoinducers (AIs) that coordinate gene expression within a population.59 At high cell numbers of E. coli relative to P. aeruginosa, sufficiently high accumulation of indole may aid in survival and growth of E. coli in presence of P. aeruginosa.
Interestingly, for co-cultures comprised of small initial cell numbers, i.e., ∼50 P. aeruginosa cells and a similar number of E. coli cells (ratio ∼ 1:1), E. coli first grows for 6 hours (to ∼2700 cells), followed by growth arrest for 4 hours and then cell death such that the E. coli cell number after 16 hours is ∼1000 cells. Here, the growth arrest and subsequent death of E. coli occurring after 10 hours may be due to the higher cell number of P. aeruginosa (∼6500) compared to E. coli (∼2500) at that time, which leads to E. coli lysis due to accumulation of sufficiently high amounts of toxic metabolites such as pyocyanin. However, the production of indole by E. coli counters the effects of pyocyanin,8 which prevents the complete eradication of the E. coli population. In the other two cases in which the initial cell number of P. aeruginosa was lower than that of E. coli (90P/190E and 30P/850E), TLFM images show that E. coli does not lyse, as expected. This increased viability may be due to higher production of indole by relatively larger number of E. coli cells and the lower production of pyocyanin production by smaller number of P. aeruginosa cells, which leads to the survival and growth arrest for E. coli in ∼6 h. In addition, the doubling time of P. aeruginosa increases from ∼50 minutes in monomicrobial cultures to ∼109 minutes (30P/850E experiment) and P. aeruginosa does not plateau after 16 hours as in other cases due to the increased doubling time. This observation is in agreement with recent literature that describes growth inhibition of P. aeruginosa in presence of metabolites such as indole produced by E. coli.8 Finally, the P. aeruginosa time–kill curve plateaus differently (760P/830E experiment) due to the higher initial cell numbers of E. coli, which affects the growth of P. aeruginosa more significantly. Overall, these results show that bacterial growth depends on interactions between the different microbes, which in turn depend on the ratio as well as the total of the initial number of cells of the different species.
Dynamics of interaction between P. aeruginosa and K. pneumoniae (absence of antimicrobials)
An improved understanding of the dynamics of interaction between P. aeruginosa and K. pneumoniae is of key interest because several urinary tract infections (UTIs) are known to be polymicrobial communities involving strains of these two species.60 Moreover, co-cultures of these bacteria also exist on the perinea of males with spinal cord injuries.61 So in the second set of co-culture experiments, we employed the microfluidic platform to monitor changes in cell numbers using TLFM in mixed cultures of P. aeruginosa and K. pneumoniae expressing RFP and GFP, respectively (Fig. 3). Mixed cultures were prepared using a similar method to that used for the P. aeruginosa and E. coli co-cultures (Fig. S2†). In experiments with low initial cell numbers of P. aeruginosa (<200), we observed that the population of K. pneumoniae increased to a threshold value (∼6700 cells), independent of the ratio of the initial cell numbers, followed by growth arrest after 6–8 hours. However, in experiments with initial cell numbers of P. aeruginosa >200, we observe almost complete cell lysis of K. pneumoniae after 16 hours irrespective of the initial cell numbers of K. pneumoniae.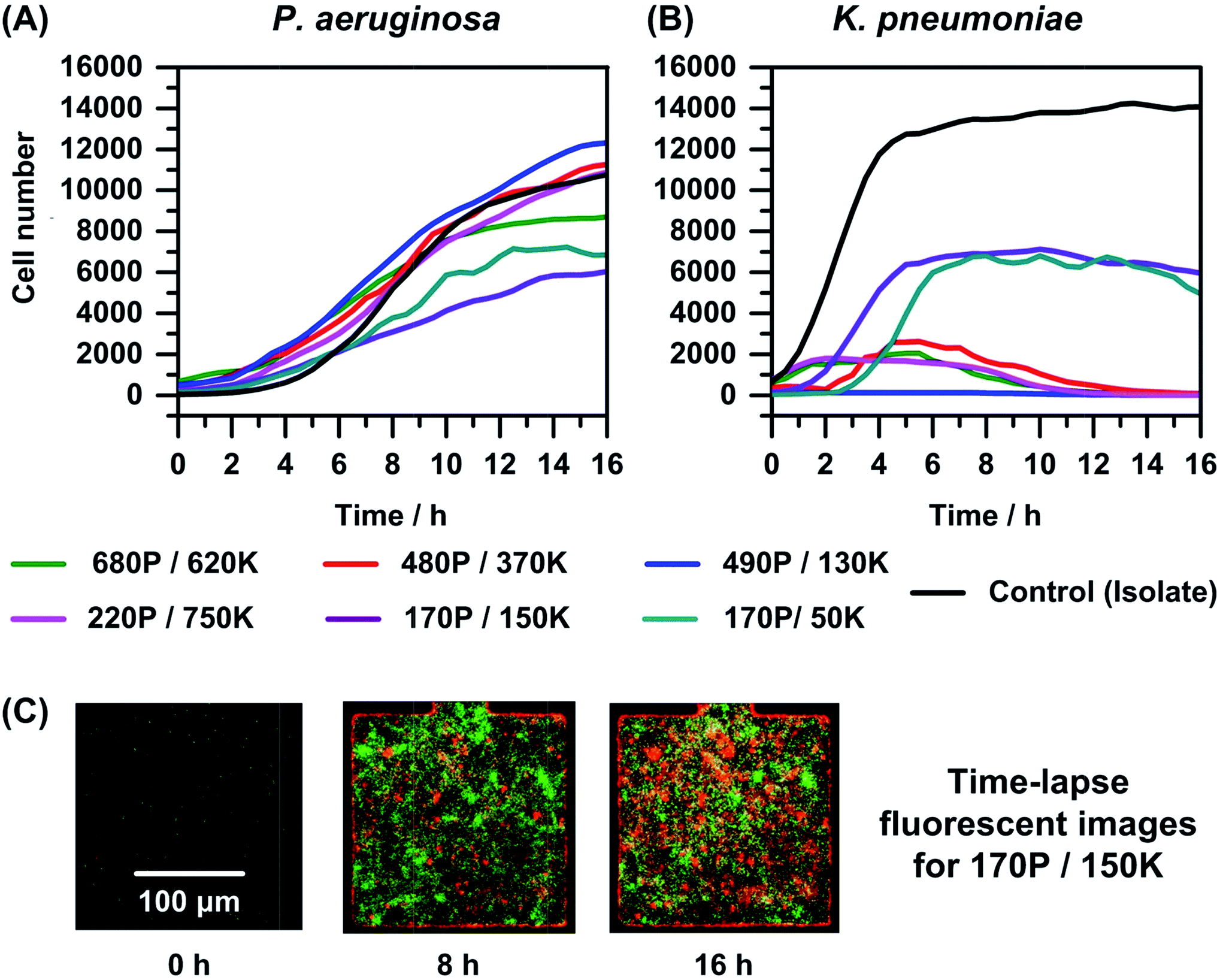 | ||
| Fig. 3 On-chip real-time monitoring of the interaction between P. aeruginosa (P) and K. pneumoniae (K) in the absence of antimicrobials. The plot represents growth curves for (A) P. aeruginosa and (B) K. pneumoniae for different starting cell numbers. Cell growth and death were monitored by counting cells in each well, every 30 minutes, over a period of 16 h. For the control curve (isolate), the initial cell numbers for P. aeruginosa and K. pneumoniae were 210 and 140. The curves are generated from data points that represent the mean of the measurements from three experiments; but we depict only the guide-lines between the data points for purposes of clarity. (C) Time-lapse fluorescent images for the case of co-culture comprising 170 P. aeruginosa cells (expressing red fluorescent protein or RFP) and 150 K. pneumoniae cells (expressing green fluorescent protein or GFP) initially (0 h). | ||
We hypothesize that the different responses of K. pneumoniae for different cell numbers are due to interspecies interactions. For co-cultures involving small numbers of cells, the concentration of AIs released by the bacteria is too low to elicit a response from the nearby bacteria. However, when a critical cell number of bacteria is present, the surrounding bacteria are able to sense and respond to AIs by upregulating transcriptional activators (i.e., R protein), thereby activating the process of quorum sensing.62 We conjecture that quorum sensing in this particular co-culture occurs for cell numbers of P. aeruginosa above 200 per well, which corresponds to a concentration of ∼108 cells per mL. The outcome of this process is growth arrest after 6–8 h and eventually lysis of K. pneumoniae. Our results suggest that K. pneumoniae likely requires more than 3000 cells (1.2 × 109 cells per mL) to survive in the presence of P. aeruginosa, as shown in Fig. 4. Overall, these results for co-culture experiments involving P. aeruginosa and K. pneumoniae show that growth dynamics is influenced by bacterial interactions, which in turn is largely dependent on the initial number of P. aeruginosa cells, but independent of the ratio of their initial cell numbers. Interestingly, this behavior is different than that observed for co-cultures of P. aeruginosa and E. coli.
 | ||
| Fig. 4 On-chip real-time monitoring of the interaction between P. aeruginosa and E. coli (co-culture) in the presence of different concentrations of tobramycin (0, 2, 4, 8, 16 μg mL−1). For comparison, similar plots for isolate cultures (dash-dotted lines) are also shown. The plot represents growth or time–kill curves for (A) P. aeruginosa and (B) E. coli. Cell growth and death were monitored by counting cells in each well, every 30 minutes, over a period of 16 h. Each data point represents the mean of the measurements from three experiments; but we depict only the guide-lines between the data points for purposes of clarity. We also plot the natural logarithm of cell numbers (N) normalized to initial cell numbers (N0), i.e., ln(N/N0), on the Y-axis, which enables comparison between experiments with differing cell numbers. Some of the growth curves do not completely extend to the 16 hour time point. Some of the curves are not plotted for higher values because accurate measurement of the number of cells at these high cell densities is inaccurate, and not relevant for interpretation. | ||
In summary, the data from two sets of polymicrobial co-culture experiments described above demonstrate significant differences in the growth dynamics of bacteria. Moreover, they showed that growth dynamics are dependent upon both the absolute and relative initial cell numbers of bacterial species in polymicrobial cultures. Through the combined use of TLFM and a microfluidic platform comprised of arrays of wells, we were able to resolve differences in growth dynamics (growth/stasis/lysis) over different time intervals, information that would be hard to obtain with conventional methods that rely on end-point assays.
Antimicrobial tolerance in polymicrobial cultures containing P. aeruginosa
P. aeruginosa is one of the primary pathogens found in many polymicrobial infections of humans.10 We performed AST against P. aeruginosa in co-cultures with E. coli (Fig. 4) and with K. pneumoniae (Fig. 5), as well as for co-cultures in which all three pathogens are present. In each experiment, the total initial cell number was set at ∼100 to 300 cells, which corresponds to a cell density of ∼108 cells per mL, similar to values published in literature for antimicrobial susceptibility testing.63,64 All these experiments were initiated with an approximately equal number of total cells to avoid inoculum effects, which is a phenomena that describes a significant increase in the MIC of an antimicrobial when the number of organisms inoculated (initial cell numbers) is increased.65 In addition, inoculum effects do not generally occur with aminoglycosides such as tobramycin against Pseudomonas species; therefore, increases in the MICs against P. aeruginosa described later in this section cannot be attributed to slight changes in total initial cell numbers.66 | ||
| Fig. 5 On-chip real-time monitoring of the interaction between P. aeruginosa and K. pneumoniae (co-culture) in the presence of different concentrations of tobramycin (0, 2, 4, 8, 16 μg mL−1). For comparison, similar plots for isolate cultures (dash-dotted lines) are also shown. The plot represents growth or time–kill curves for (A) P. aeruginosa and (B) K. pneumoniae. Cell growth and death were monitored by counting cells in each well, every 30 minutes, over a period of 16 h. Each data point represents the mean of the measurements from three experiments; but we depict only the guide-lines between the data points for purposes of clarity. We also plot the natural logarithm of cell numbers (N) normalized to initial cell numbers (N0), i.e., ln(N/N0), on the Y-axis, which enables comparison between experiments with differing cell numbers. Some of the curves are not plotted for higher values because accurate measurement of the number of cells at these high cell densities is inaccurate, and not relevant for interpretation. | ||
Fig. 4 shows the growth or time–kill curves for co-cultures of P. aeruginosa with E. coli, in the presence of varying concentrations of tobramycin. For reference, we also include the time–kill curves for isolate cultures in the same plot (dash-dotted lines). Cell growth and death were monitored by counting cells in each well, every 30 minutes, over a period of 16 hours. Each data point was obtained by taking the mean of the measurements from three experiments. For clarity we depict only the lines that connect the data-points in Fig. 4 and 5. We also plot the natural logarithm of cell numbers (N) normalized to initial cell numbers (N0), i.e., ln(N/N0), on the Y-axis, which enables comparison between experiments with differing cell numbers.
In the case of co-culturing P. aeruginosa with E. coli (Fig. 4(B)), we observed that growth of E. coli is inhibited (lower cell numbers after 16 hours) compared to its growth in an isolate culture (0 and 2 μg mL−1 curves). This observation is consistent with the results depicted in Fig 2(B), which compare co-culture growth curves in the absence of antimicrobials. The observed MICs of tobramycin against E. coli in an isolate culture or in co-culture with P. aeruginosa are similar, ranging from 2 to 4 μg mL−1.
In contrast, the growth of P. aeruginosa, when in co-culture with E. coli in the presence of 0, 2, 4 μg mL−1 of tobramycin, is enhanced compared to its growth in isolate cultures under identical conditions (Fig 4(A)). This observation is counter-intuitive as one would expect similar or more probably lower cell growth in co-culture compared to isolate, because the cell growth was slightly inhibited in similar co-cultures in the absence of antimicrobials (Fig. 2(A)). This counter-intuitive observation can be attributed to the previously reported enhanced virulence and/or antimicrobial resistance of P. aeruginosa in co-cultures with different species or mixed cultures of different strains.6,10 In the experiment shown in Fig. 4(A) the MIC increases to 16 μg mL−1 in co-culture compared to 4 μg mL−1 in isolate. In repeats of these experiments, we consistently observed a 2- to 4-fold increase in MIC of tobramycin against P. aeruginosa in co-culture vs. isolate.
The growth or time–kill curves for co-cultures of P. aeruginosa with K. pneumoniae, in the presence of varying concentrations of tobramycin are shown in Fig. 5. We observed that the growth or time–kill curves for K. pneumoniae in the presence of the antimicrobial agent (Fig. 5(B)) are similar for co-culture and isolates. This observation is consistent with the similar growth curves observed when comparing isolate and co-cultures with ∼1:1 initial cell numbers in the absence of tobramycin (Fig. 3(B)). The MIC value against K. pneumoniae was unchanged in co-culture compared to isolate (16 μg mL−1), which is consistent with observations in similar co-culture experiments with different initial cell numbers (plots not shown). In contrast, we observed enhanced growth of P. aeruginosa in co-culture compared to isolate (Fig. 5(A)), similar to the behavior observed in co-culture experiments of P. aeruginosa with E. coli (Fig. 4(A)). In the experiment shown in Fig. 5(A), we observed that in co-culture the MIC increases to 16 μg mL−1 compared to 4 μg mL−1 in isolate. In repeats of these experiments, we observed similar 2- to 4-fold increases in MICs of tobramycin against P. aeruginosa for co-cultures compared to isolates.
In addition to experiments with co-culture comprising two species, we also determined the MIC of tobramycin against P. aeruginosa in co-cultures comprising three species, so P. aeruginosa, with E. coli and K. pneumoniae. The MIC against P. aeruginosa in this co-culture increased 4- to 8-fold compared to isolate. We also observed that supra-lethal concentrations of tobramycin (as high as 256 μg mL−1) failed to cause complete eradication of all three species. Note that in the studies reported here, two of the strains (E. coli and K. pneumoniae) expressed the same color (green), which made it challenging to study the interaction between these two species and determine species-specific MIC. This issue can be conveniently addressed by using strains expressing different colored proteins (e.g., yellow or blue) and appropriate filters for these colors, thus enabling the determination of MIC for all the strains involved.
The observed increase in MIC values against P. aeruginosa in the three sets of co-culture experiments described above, demonstrates its increased antimicrobial tolerance in co-cultures. Three mechanisms are known to cause antimicrobial resistance: (1) antimicrobials are prevented from interacting with target sites, (2) efflux of the antimicrobial from the bacterial cells before reaching target sites of attack, and/or (3) direct destruction or modification of the antimicrobial molecule.67 Antimicrobial resistance mediated by the first mechanism can occur in scenarios where the metabolic activity of bacteria is lowered in co-cultures compared to that in isolate,68 which in turn leads to lower activity of the antimicrobial target sites. This lowering of metabolic activity may result from a need to conserve energy due to the competition for available nutrients in co-cultures. In fact, bacteria growth in conditions of limited nutrients can initiate a mechanism known as stringent response, which leads to growth arrest and subsequent inactivity of the antimicrobial target sites (e.g., binding elements such as ribosomal RNA), thereby resulting in increased antimicrobial tolerance.68 With respect to the second mechanism, interspecies communication in bacteria is known to change gene expression patterns, which may cause efflux of antimicrobial molecules, leading to increased antimicrobial resistance. For example, the efflux pump genes in P. aeruginosa can be up-regulated in mixed co-cultures.69 The third mechanism of antimicrobial resistance involving the destruction or modification of antimicrobial molecules may be different in isolate versus co-cultures due to differences in the production of molecules that can modify antimicrobials. Bacterial interactions may result in the production of distinct metabolites that differ from those produced in isolate cultures. Indeed, this is an exciting area of study, because the identity of several of these chemical agents is yet to be resolved.23 Nevertheless, known metabolites that are specific to certain pathogens can be monitored. For example, in the case of P. aeruginosa, the increased production of pyocyanin observed in co-cultures with gram positive bacteria (such as S. aureus) is often associated with increased virulence,23,70 whereas a decreased amount of pyocyanin is produced in the presence of a gram negative bacterial strain such as E. coli ZK126.8 Furthermore, in co-cultures of E. coli and P. aeruginosa, increased production of a different metabolite (indole) aids in survival of E. coli as discussed above.8
Interestingly, when we performed identical experiments using amikacin instead of tobramycin as the antimicrobial agent, we did not observe significant changes between the MICs and the time–kill curves obtained for isolate vs. polymicrobial conditions (data not shown). A possible reason for this observation is that amikacin is less prone to inactivation by P. aeruginosa, e.g., amikacin has a low tendency to be degraded by the enzymes secreted by the bacteria.71,72 Hence, amikacin is similarly effective against P. aeruginosa irrespective of being present as an isolate or in co-culture. This high resistance of amikacin against bacterial inactivation is one of the prime reasons for amikacin being used as a last resort to treat infections involving P. aeruginosa.71
Conclusions
In this work, we reported the utility of a microfluidic approach for quantification of interactions between P. aeruginosa, E. coli, and K. pneumoniae in the presence and absence of antimicrobials. This culture-independent method allows for the study of species-specific antimicrobial susceptibility and real-time monitoring of bacterial interaction. The microfluidic approach reported here has several advantages over conventional methods (e.g., broth dilution), including shorter experiment times by obviating the need to pre-culture the bacteria and more quantitative data by using time–kill curves as opposed to end-point assay. Also, this approach of directly enumerating the cells by real-time visual observation is simpler and less expensive compared to qT-RFLP, and hence has potential for translation to clinical settings.It is well known that in mixed populations of microbes, the growth dynamics of the individual strains or species depends on the absolute and relative number of initial cells. Additionally, and more importantly, this interaction among the different types of microbes affects their antimicrobial susceptibility and it complicates AST. The antimicrobial resistance of a certain microbe is influenced by several factors, including phenotype of all the microbes present and the antimicrobial used. The microfluidic approach reported here allows for systematic study of the effects of these different factors on antimicrobial resistance. Such studies may also enhance understanding of the different mechanisms causing antimicrobial resistance. While we did not study the influence of phenotype variation on antimicrobial susceptibility, the microfluidic approach presented here is suitable for such studies.
The microfluidic approach reported here relies on TLFM analysis of on-chip cell cultures comprised of genetically modified bacteria, an approach that is not appropriate to analyze clinical samples. Additionally the use of genetic modifications may affect the growth dynamics. Hence in ongoing work, we are exploring the use of optical dyes for long-term monitoring of the growth dynamic of wild-type bacteria. This approach of using genetically modified bacteria, however, can still be used to uncover the different mechanisms governing polymicrobial interactions.
With further development, microfluidic platforms such as those used in this study may potentially be used as a clinical diagnostic platform for AST in case of polymicrobial infections. One area of development that is required for clinical translation is the ability to distinguish between different species without genetically modifying the bacteria. Approaches based on differences in morphological characteristics and/or motility behaviour may enable this distinction. Another important aspect to consider for clinical translation, especially for treating polymicrobial infections, is to minimize the effect of the relative starting cell numbers on MIC values. To account for this effect, multiple samples (with potentially different relative cell numbers) can be tested against the same antimicrobial concentration. Here the microfluidic approach is advantageous because of the multiplexing capability that enables testing of many conditions with small sample volumes.
Typically, precise determination of MICs is sufficient to predict the effectiveness of specific antimicrobials against specific infections; a microfluidic assay can rapidly provide such data of immediate clinical utility (hours) compared to the longer time (days) needed by the methods currently used in most clinical settings. Looking forward, the in vitro AST results obtained from the microfluidic platform (time–kill curves) can be utilized to perform pharmacodynamics/pharmacokinetics (PD/PK) modelling, a more advanced but rarely used method to predict in vivo antimicrobial dosing regimen for humans.73 Furthermore, the ability to perform long-term polymicrobial cell studies in multiplexed fashion on chip may also have significant potential in other areas of microbiology, e.g., study of the responses of mixed bacterial populations to external stresses, screening of drug candidates, optimization of polymicrobial co-culture conditions for biofuel production, etc.
Acknowledgements
We acknowledge financial support from the National Science Foundation under awards CMMI 03-28162 and CMMI 07-49028 to Nano-CEMMS; a Nano Science & Engineering Center (NSEC) on Nanomanufacturing for PJAK. We also thank Dr Ashtamurthy Pawate, Kori Dunn for useful discussions, and Dr Arnab Mukherjee for his assistance in the early progress of the project. Finally, we thank Prof. Eric Triplett from the University of Florida and Prof. Robert Shanks from the University of Pittsburgh for kindly providing fluorescently labelled bacteria.References
- P. D. Straight and R. Kolter, Annu. Rev. Microbiol., 2009, 63, 99–118 CrossRef CAS PubMed.
- S. A. West, A. S. Griffin, A. Gardner and S. P. Diggle, Nat. Rev. Microbiol., 2006, 4, 597–607 CrossRef CAS PubMed.
- K. A. Brogden, J. M. Guthmiller and C. E. Taylor, Lancet, 2005, 365, 253–255 CrossRef.
- K. A. Brogen and J. M. Guthmiller, Polymicrobial diseases, ASM Press, 2002 Search PubMed.
- D. Kiani, E. L. Quinn, K. H. Burch, T. Madhavan, L. D. Saravolatz and T. R. Neblett, J. Am. Med. Assoc., 1979, 242, 1044–1047 CrossRef CAS.
- G. B. Rogers, L. R. Hoffman, M. Whiteley, T. W. V. Daniels, M. P. Carroll and K. D. Bruce, Trends Microbiol., 2010, 18, 357–364 CrossRef CAS PubMed.
- M. P. Weinstein, L. B. Reller and J. R. Murphy, Diagn. Microbiol. Infect. Dis., 1986, 5, 185–196 CrossRef CAS.
- W. Chu, T. R. Zere, M. M. Weber, T. K. Wood, M. Whiteley, B. Hidalgo-Romano, E. Valenzuela and R. J. C. McLean, Appl. Environ. Microbiol., 2012, 78, 411–419 CrossRef CAS PubMed.
- M. E. Hibbing, C. Fuqua, M. R. Parsek and S. B. Peterson, Nat. Rev. Microbiol., 2010, 8, 15–25 CrossRef CAS PubMed.
- A. Korgaonkar, U. Trivedi, K. P. Rumbaugh and M. Whiteley, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 1059–1064 CrossRef CAS PubMed.
- F. E. McKenzie, J. Clin. Epidemiol., 2006, 59, 760–761 CrossRef CAS PubMed.
- S. P. Brown, M. E. Hochberg and B. T. Grenfell, Trends Microbiol., 2002, 10, 401–405 CrossRef CAS.
- C. Cillóniz, S. Ewig, M. Ferrer, E. Polverino, A. Gabarrús, J. Puig de la Bellacasa, J. Mensa and A. Torres, Crit. Care, 2011, 15(5), R209 CrossRef PubMed.
- A. K. Wessel, L. Hmelo, M. R. Parsek and M. Whiteley, Nat. Rev. Microbiol., 2013, 11, 337–348 CrossRef CAS PubMed.
- R. P. Ryan and J. M. Dow, Microbiology, 2008, 154, 1845–1858 CrossRef CAS PubMed.
- P. Gupta, G. D. Kumhar, G. Kaur and V. G. Ramachandran, J. Paediatr. Child Health, 2005, 41, 365–368 CrossRef PubMed.
- A. R. Marra, G. M. L. Bearman, R. P. Wenzel and M. B. Edmond, BMC Infect. Dis., 2005, 5, 94 CrossRef PubMed.
- S. Y. Park, K. H. Park, K. M. Bang, Y. P. Chong, S. H. Kim, S. O. Lee, S. H. Choi, J. Y. Jeong, J. H. Woo and Y. S. Kim, J. Infect., 2012, 65, 119–127 CrossRef PubMed.
- S. B. Levy and B. Marshall, Nat. Med., 2004, 10, S122–S129 CrossRef CAS PubMed.
- S. P. Diggle, Microbiology, 2010, 156, 3503–3512 CrossRef CAS PubMed.
- D.-C. Oh, P. R. Jensen, C. A. Kauffman and W. Fenical, Bioorg. Med. Chem., 2005, 13, 5267–5273 CrossRef CAS PubMed.
- D.-C. Oh, C. A. Kauffman, P. R. Jensen and W. Fenical, J. Nat. Prod., 2007, 70, 515–520 CrossRef CAS PubMed.
- E. A. Shank and R. Kolter, Curr. Opin. Microbiol., 2009, 12, 205–214 CrossRef CAS PubMed.
- C. Riedele and U. Reichl, J. Antimicrob. Chemother., 2011, 66, 138–145 CrossRef CAS PubMed.
- O. M. El-Halfawy and M. A. Valvano, PLoS One, 2013, 8(7), e68874 CAS.
- F. Heilmann, Infection, 1993, 21, 187–190 CrossRef CAS.
- W. R. Heizmann, F. Heilmann, B. Egeler and H. Werner, Infection, 1990, 18, 117–121 CrossRef CAS.
- H. Werner, W. R. Heizmann, G. Freuer, N. Mitinis and F. Heilmann, J. Antimicrob. Chemother., 1989, 24, 55–61 CrossRef CAS PubMed.
- O. Lazcka, F. J. D. Campo and F. X. Muñoz, Biosens. Bioelectron., 2007, 22, 1205–1217 CrossRef CAS PubMed.
- R. L. White, D. S. Burgess, M. Manduru and J. A. Bosso, Antimicrob. Agents Chemother., 1996, 40, 1914–1918 CAS.
- J. K. Schmidt, B. König and U. Reichl, Biotechnol. Bioeng., 2007, 96, 738–756 CrossRef CAS PubMed.
- C. Riedele and U. Reichl, Eng. Life Sci., 2012, 12, 188–197 CrossRef CAS PubMed.
- Y. Zheng, J. Nguyen, Y. Wei and Y. Sun, Lab Chip, 2013, 13, 2464–2483 RSC.
- J. H. Yeon and J.-K. Park, BioChip J., 2007, 1, 17–27 Search PubMed.
- J. Wu, X. Wu and F. Lin, Lab Chip, 2013, 13, 2484–2499 RSC.
- T. P. Lagus and J. F. Edd, J. Phys. D: Appl. Phys., 2013, 46, 114005 CrossRef.
- J. Park, A. Kerner, M. A. Burns and X. N. Lin, PLoS One, 2011, 6, e17019 CAS.
- S. Park, D. Kim, R. J. Mitchell and T. Kim, Lab Chip, 2011, 11, 2916–2923 RSC.
- J. Q. Boedicker, M. E. Vincent and R. F. Ismagilov, Angew. Chem., Int. Ed., 2009, 48, 5908–5911 CrossRef CAS PubMed.
- C. H. Chen, Y. Lu, M. L. Y. Sin, K. E. Mach, D. D. Zhang, V. Gau, J. C. Liao and P. K. Wong, Anal. Chem., 2010, 82, 1012–1019 CrossRef CAS PubMed.
- K. Churski, T. S. Kaminski, S. Jakiela, W. Kamysz, W. Baranska-Rybak, D. B. Weibel and P. Garstecki, Lab Chip, 2012, 12, 1629–1637 RSC.
- N. J. Cira, J. Y. Ho, M. E. Dueck and D. B. Weibel, Lab Chip, 2012, 12, 1052–1059 RSC.
- J. Y. Ho, N. J. Cira, J. A. Crooks, J. Baeza and D. B. Weibel, PLoS One, 2012, 7, e41245 CAS.
- M. Kalashnikov, J. C. Lee, J. Campbell, A. Sharon and A. F. Sauer-Budge, Lab Chip, 2012, 12, 4523–4532 RSC.
- P. Sun, Y. Liu, J. Sha, Z. Zhang, Q. Tu, P. Chen and J. Wang, Biosens. Bioelectron., 2011, 26, 1993–1999 CrossRef CAS PubMed.
- R. Mohan, A. Mukherjee, S. E. Sevgen, C. Sanpitakseree, J. Lee, C. M. Schroeder and P. J. A. Kenis, Biosens. Bioelectron., 2013, 49, 118–125 CrossRef CAS PubMed.
- J. Choi, Y.-G. Jung, J. Kim, S. Kim, Y. Jung, H. Na and S. Kwon, Lab Chip, 2013, 13, 280–287 RSC.
- A. B. Theberge, F. Courtois, Y. Schaerli, M. Fischlechner, C. Abell, F. Hollfelder and W. T. S. Huck, Angew. Chem., Int. Ed., 2010, 49, 5846–5868 CrossRef CAS PubMed.
- J. H. Jorgensen and M. J. Ferraro, Clin. Infect. Dis., 2009, 49, 1749–1755 CrossRef CAS PubMed.
- Y. Xia and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 550–575 CrossRef CAS.
- A. Mukherjee, J. Walker, K. B. Weyant and C. M. Schroeder, PLoS One, 2013, 8, e64753 CAS.
- M. K. Chelius and E. W. Triplett, Appl. Environ. Microbiol., 2000, 66, 783–787 CrossRef CAS.
- R. M. Q. Shanks, D. E. Kadouri, D. P. MacEachran and G. A. O'Toole, Plasmid, 2009, 62, 88–97 CrossRef CAS PubMed.
- R. Mohan, B. R. Schudel, A. V. Desai, J. D. Yearsley, C. A. Apblett and P. J. A. Kenis, Sens. Actuators, B, 2011, 160, 1216–1223 CrossRef CAS PubMed.
- T. Thorsen, S. J. Maerkl and S. R. Quake, Science, 2002, 298, 580–584 CrossRef CAS PubMed.
- J. E. Keymer, P. Galajda, C. Muldoon, S. Park and R. H. Austin, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 17290–17295 CrossRef CAS PubMed.
- N. C. Shaner, P. A. Steinbach and R. Y. Tsien, Nat. Methods, 2005, 2, 905–909 CrossRef CAS PubMed.
- S. S. Baron and J. J. Rowe, Antimicrob. Agents Chemother., 1981, 20, 814–820 CrossRef CAS.
- D. T. Hughes and V. Sperandio, Nat. Rev. Microbiol., 2008, 6, 111–120 CrossRef CAS PubMed.
- S. M. Jacobsen, D. J. Stickler, H. L. T. Mobley and M. E. Shirtliff, Clin. Microbiol. Rev., 2008, 21, 26–59 CrossRef CAS PubMed.
- D. S. Gilmore, D. G. Schick and J. Z. Montgomerie, J. Clin. Microbiol., 1982, 16, 865–867 CAS.
- T. R. De Kievit and B. H. Iglewski, Infect. Immun., 2000, 68, 4839–4849 CrossRef CAS.
- J. B. Patel, F. C. Tenover, J. D. Turnidge and J. H. Jorgensen, in Manual of Clinical Microbiology, American Society of Microbiology, 10th edn, 2011 Search PubMed.
- J. D. Turnidge, M. J. Ferraro and J. H. Jorgensen, in Manual of Clinical Microbiology, American Society of Microbiology, 10th edn, 2011 Search PubMed.
- E. Bidlas, T. Du and R. J. W. Lambert, Int. J. Food Microbiol., 2008, 126, 140–152 CrossRef CAS PubMed.
- I. Brook, Rev. Infect. Dis., 1989, 11, 361–368 CrossRef CAS PubMed.
- G. D. Wright, Adv. Drug Delivery Rev., 2005, 57, 1451–1470 CrossRef CAS PubMed.
- D. Nguyen, A. Joshi-Datar, F. Lepine, E. Bauerle, O. Olakanmi, K. Beer, G. McKay, R. Siehnel, J. Schafhauser, Y. Wang, B. E. Britigan and P. K. Singh, Science, 2011, 334, 982–986 CrossRef CAS PubMed.
- K. Duan, C. Dammel, J. Stein, H. Rabin and M. G. Surette, Mol. Microbiol., 2003, 50, 1477–1491 CrossRef CAS.
- N. M. Vega, K. R. Allison, A. N. Samuels, M. S. Klempner and J. J. Collins, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14420–14425 CrossRef CAS PubMed.
- L. Gonzalez and J. Spencer, Am. Fam. Phys., 1998, 58, 1811–1820 Search PubMed.
- K. Poole, Antimicrob. Agents Chemother., 2005, 49, 479–487 CrossRef CAS PubMed.
- R. R. Regoes, C. Wiuff, R. M. Zappala, K. N. Garner, F. Baquero and B. R. Levin, Antimicrob. Agents Chemother., 2004, 48, 3670–3676 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra04092b |
| This journal is © The Royal Society of Chemistry 2015 |