
PtAu alloy nanoparticles supported on thermally expanded graphene oxide as a catalyst for the selective oxidation of glycerol†
Abstract
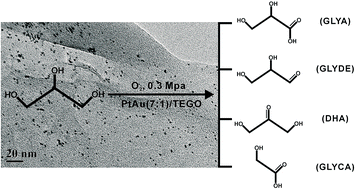
Thermally expanded graphene oxide (TEGO) supported PtAu alloy nanoparticles with various compositions was prepared, and then characterized using a combination of atomic absorption spectroscopy, powder X-ray diffraction, transmission electron microscopy and X-ray photoelectron spectroscopy. These catalysts were evaluated for the aerobic oxidation of glycerol in base-free aqueous solution. The results showed that PtAu(7 : 1)/TEGO exhibited the optimum activity for the conversion of glycerol among all the catalysts. Glycerol conversion of 60.4% and selectivities of 53.5% glyceric acid (GLYA), 25.7% glyceraldehydes (GLYDE), 11.6% dihydroxyacetone (DHA), and 3.8% glycolic acid (GLYCA) were obtained when reacting an aqueous solution of glycerol (0.3 M, 20 mL) at 60 °C under 0.3 MPa O2 for 4 h in the presence of 0.023 g PtAu(7 : 1)/TEGO catalyst. Moreover, the reusability of PtAu(7 : 1)/TEGO was investigated, and a reaction mechanism for the oxidation of glycerol was proposed.
 Please wait while we load your content...
Please wait while we load your content...

