
Cu doped Fe3O4 magnetic adsorbent for arsenic: synthesis, property, and sorption application†
Abstract
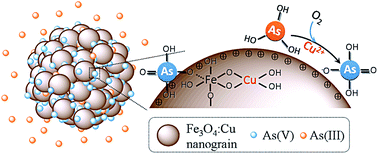
Cu doped Fe3O4 (Fe3O4:Cu) particles were synthesized and applied for arsenic adsorption. As the copper ions increase, the adsorption capacity of Fe3O4:Cu towards As(V) and As(III) increases from 7.32 to 42.90 mg g−1 and from 8.12 to 37.97 mg g−1, respectively. The incorporation of copper decreased the particle size, increased the surface area, porosity and zeta potential, leading to the increase of the adsorption sites and affinity toward negative As(V) species. More importantly, the doped copper ions catalyzed the efficient oxidation of As(III) to As(V) by O2 followed by As(V) adsorption. The Fe3O4:Cu particles also exhibited good performance toward low level arsenic removal, excellent separation, and satisfactory regeneration property. The results indicate Fe3O4:Cu particles possess great potential for both As(III) and As(V) adsorption.
 Please wait while we load your content...
Please wait while we load your content...

