DOI:
10.1039/C5RA03921E
(Paper)
RSC Adv., 2015,
5, 31104-31112
exo-Methylene-BEDT-TTF and alkene-functionalised BEDT-TTF derivatives: synthesis and radical cation salts†
Received
5th March 2015
, Accepted 25th March 2015
First published on 25th March 2015
Abstract
Hydroxymethyl-BEDT-TTF is converted by tosylation and elimination into exo-methylene-BEDT-TTF whose exocyclic double bond leads to a small increase in the oxidation potential, and from which radical cation salts with perchlorate and triiodide have been prepared, the latter showing semiconductor properties. Allyloxymethyl- and allylthiomethyl-BEDT-TTF, the first BEDT-TTF derivatives with an alkene in the side chain, have been prepared, along with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 radical cation perchlorate salt of the former, an insulator which contains a donor dication.
:2 radical cation perchlorate salt of the former, an insulator which contains a donor dication.
Introduction
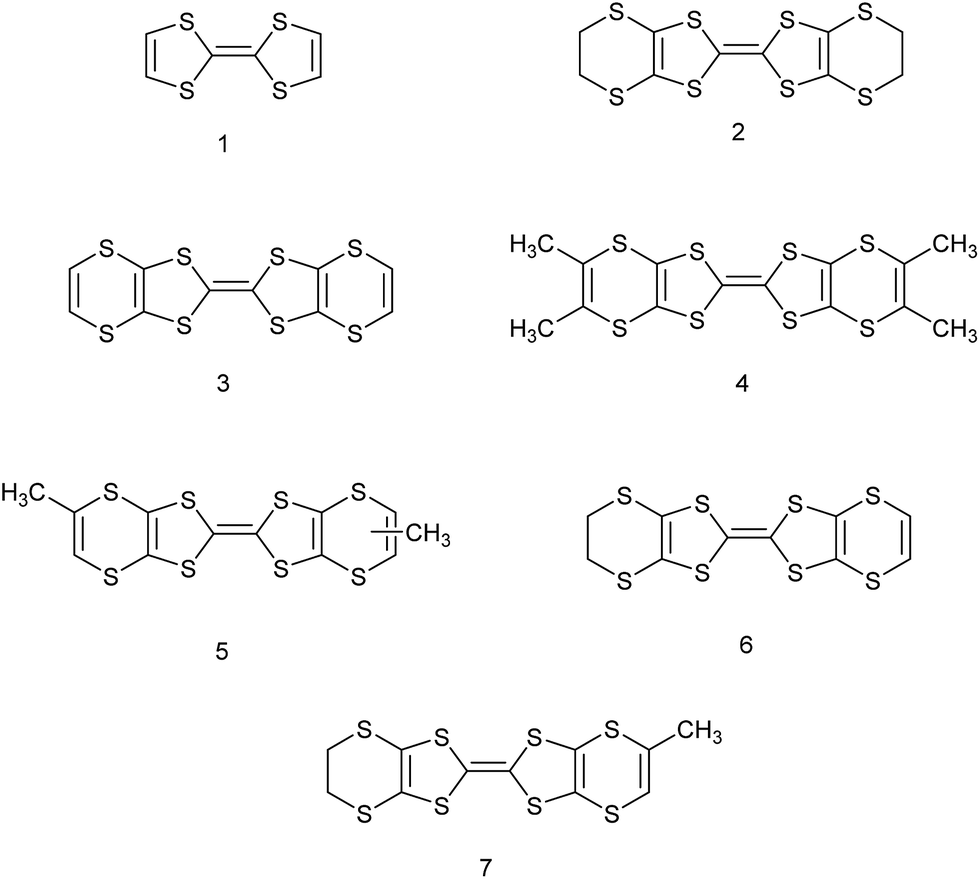
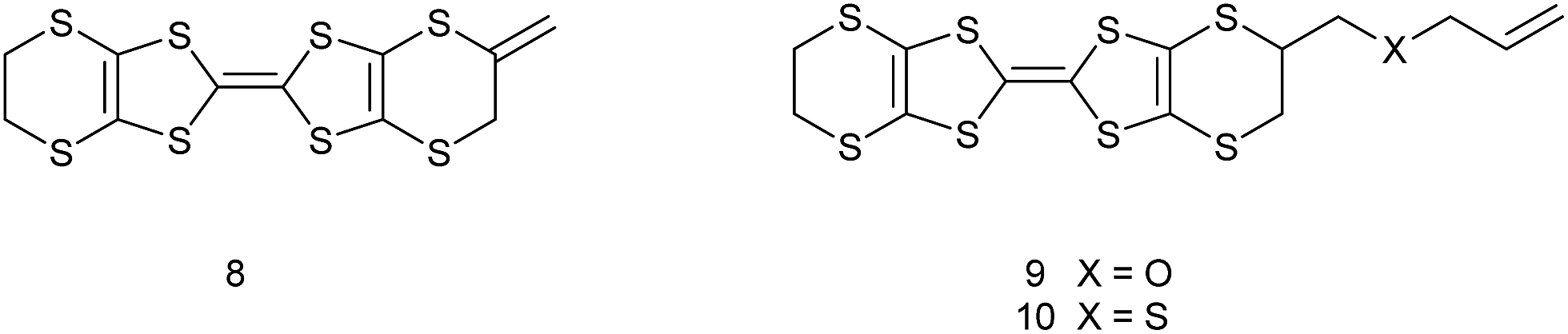
Organosulfur donors such as TTF 1 and BEDT-TTF 2 have been used as substrates for preparation of conducting materials by oxidation either by electrocrystallisation or treatment with electron-deficient acceptor substances, leading to conducting, semi-conducting and in some cases, superconducting materials.1 A wide range of substituted derivatives of these donors has been prepared and applications explored.2 There has been less interest in donor 3, known as VT, the fully unsaturated derivative of BEDT-TTF containing endocyclic double bonds at both ends of the molecule, though several salts have been prepared.3,4 Substituted derivatives of VT have been prepared including the tetra-methyl, -phenyl and -pyrid-4-yl substituted derivatives, for example 4,5 as well as the dimethyl derivative 5 which has been prepared as a mixture of cis and trans isomers.4 There are a few reports of the BEDT-TTF derivative 6 where just one dithiin ring contains an extra double bond,6,7 and some of its monosubstituted derivatives such as the methyl derivative 7.6,8 The inclusion of one or two extra double bonds raises the oxidation potential from that of BEDT-TTF by 0.06 and 0.14 V respectively.6 We have been interested in preparing donors with alkene functionality, partly because of the potential for polymerising such systems, as well as for the donors themselves as substrates for salt formation. Here we report the preparation of three new donors 8–10, one with an exocyclic double bond attached to the BEDT-TTF framework and thus a tautomer of 7, and two with an alkene group at the end of a side chain.
Discussion
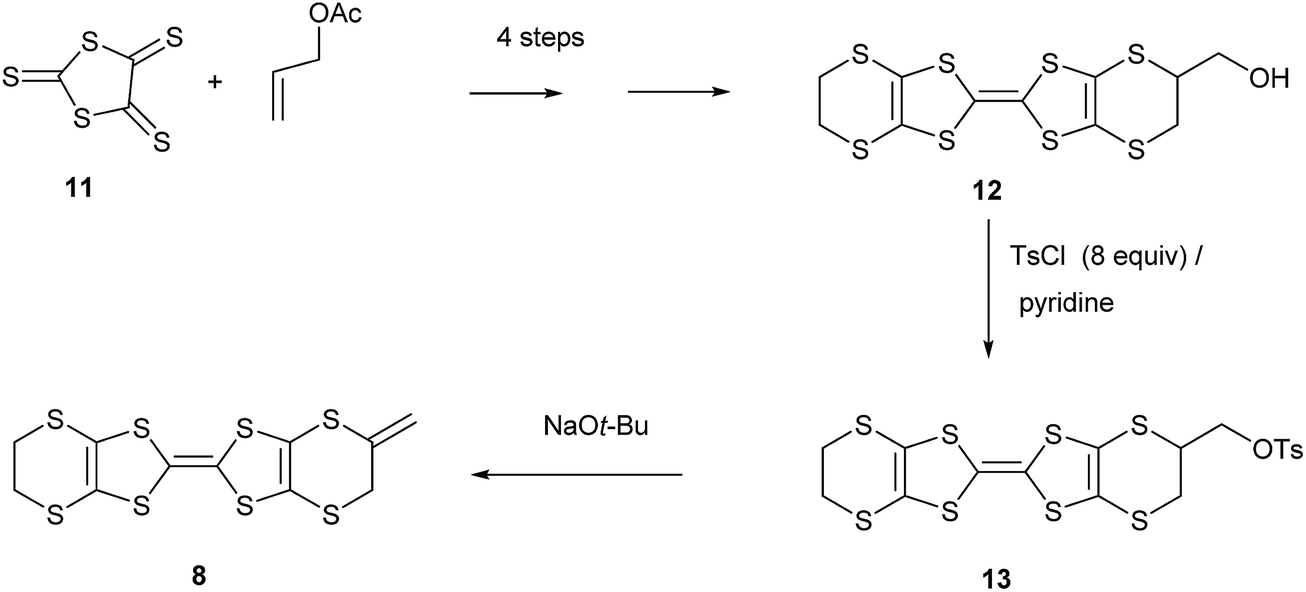
Synthesis and properties of exo-methylene-BEDT-TTF
Hydroxymethyl-BEDT-TTF 12 is readily prepared in four steps from allyl acetate and trithione 11,9 but has not been significantly exploited synthetically. We now report that conversion to its O-tosylate 139 and subsequent treatment with t-butoxide eliminates tosic acid to give exo-methylene-BEDT-TTF 8 which has an exocyclic double bond connected to the BEDT-TTF system. We were unable to isomerise 8 to the donor 7 with an exocyclic methyl group and endocyclic double bond. The 1H NMR spectrum of donor exo-methylene BEDT-TTF shows signals for the alkene hydrogens at δ: 5.13 and 5.22 ppm and for the adjacent methylene group at δ: 3.52 ppm. In contrast donor 7 clearly shows the methyl and vinyl hydrogen signals (δ: 2.11 and 6.53 ppm) from the unsaturated ring and quite a different melting point.6,8 Furthermore, the 1H NMR of the dimethyl-VT donor 5 also clearly shows the presence of the methyl and vinyl hydrogens (Scheme 1).4
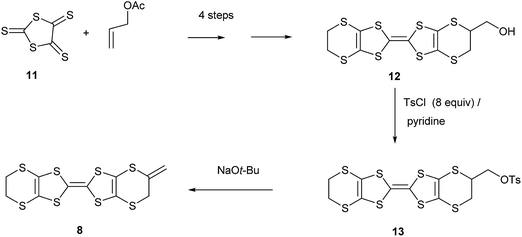 |
| | Scheme 1 | |
For the new donor 8 the consequence of conjugation of the organosulfur system with the exocyclic bond is a small increase in the first two oxidation potentials from 0.52 and 0.94 V (relative to Ag/AgCl) for BEDT-TTF to 0.56 and 0.97 V for exo-methylene-BEDT-TTF (Table 1).
Table 1 Cyclic voltammetry of donors 8, 9 and 10
| Compound |
E
1
|
E
2
|
| BEDT-TTF, 2 |
0.52 |
0.94 |
|
exo-Methylene-BEDT-TTF, 8 |
0.56 |
0.97 |
| Allyloxymethyl-BEDT-TTF, 9 |
0.54 |
0.94 |
| Allylthiomethyl-BEDT-TTF, 10 |
0.54 |
0.94 |
Electrocrystallisation experiments on exo-(methylene)BEDT-TTF 8 in the presence of various anions were carried out, and the most successful was that with perchlorate which gave thin black crystals of a 1:1 radical cation salt. Furthermore, diffusion of a dichloromethane solution of iodine into a benzonitrile solution of 8 led to a 2:2:1 salt of the donor monocation with triiodide and iodine which has been shown to be a semiconductor.
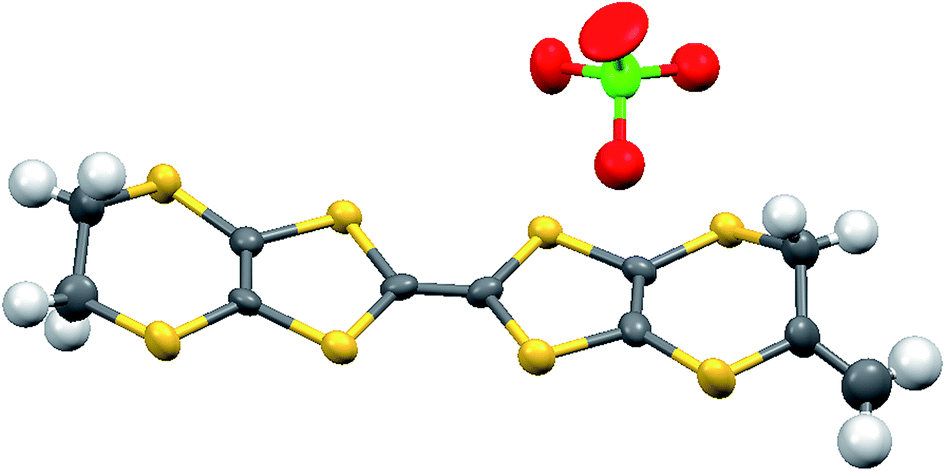
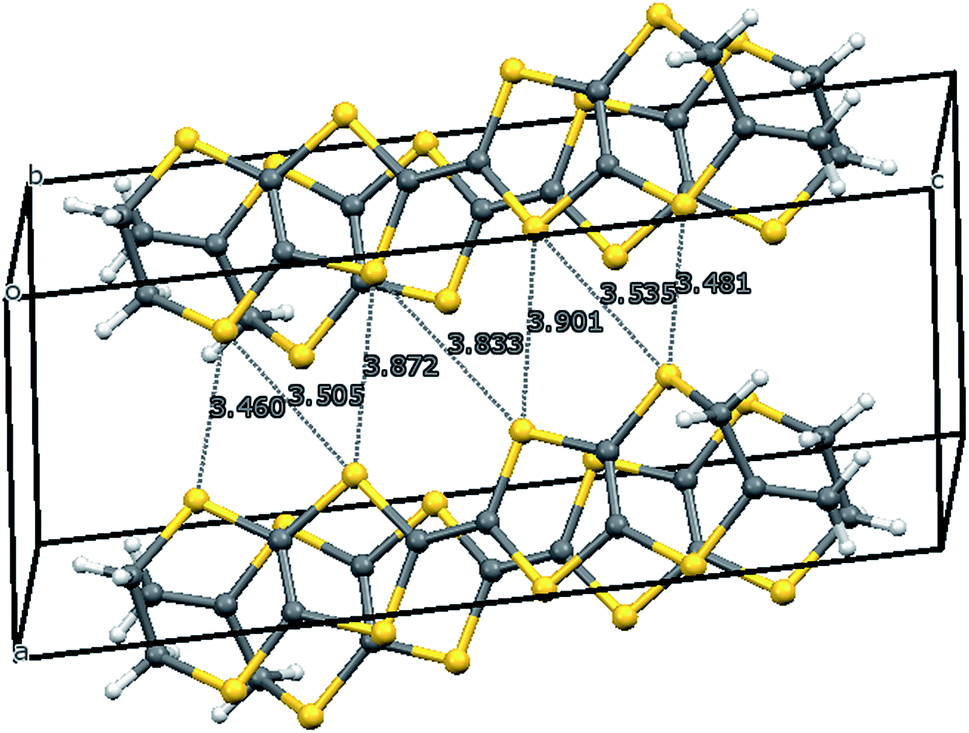
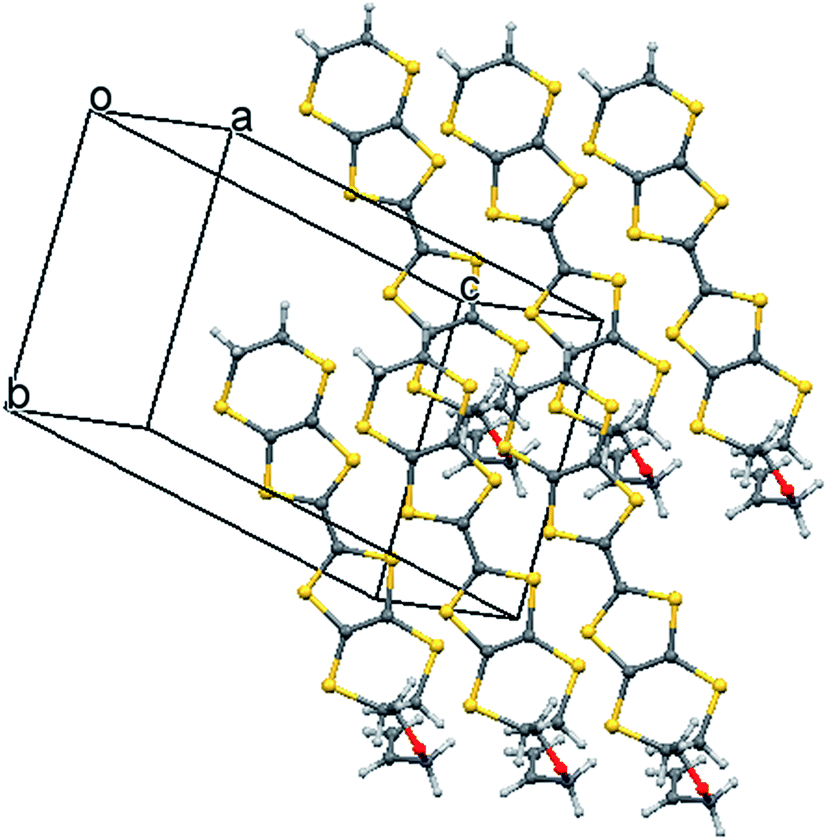
The crystal structure of the 1:1 salt with perchlorate was measured at 100 K. The crystal system is monoclinic, in space group P21/c, with one donor cation and one perchlorate anion in the asymmetric unit (Fig. 1). The crystals are twinned and there is some orientational disorder of the perchlorate ion, as well as some of the commonly observed conformational disorder in the unsubstituted dithiin ring. The donor cations are organised in centrosymmetric pairs, and these are packed in the bc plane along with the anions (Fig. 2) which isolate pairs from one another. However, there are short contacts between pairs which lie side by side in the a direction (Fig. 3). In the donor cation the effect of the exocyclic alkene is a shortening of the endocyclic bonds connecting it to its sulfur and carbon atom neighbours to 1.758(13) and 1.457(18) Å. The alkene bond length is 1.32(2) Å but the apparent shortness may be due to some slight positional disorder of the terminal methylene group. The conformation of the substituted dithiin ring is an envelope with the sp3 carbon atom at the envelope's flap. The exocyclic sp2 methylene group lies to the opposite side. Within a donor cation dimer there are four short S⋯S contacts between the centrosymmetrically related TTF moieties (3.375(4) and 3.504(5) Å), and there are four short S⋯S contacts in the range 3.460(5)–3.535(5) Å between dimers (Fig. 3), thus providing a possible route for conduction of electrons through the structure. Unfortunately, the crystals were too brittle for attachment of electrodes for conductivity measurements.
 |
| | Fig. 1 Molecular structure of the salt 8·ClO4 with anisotropic displacement parameters drawn at the 50% level. | |
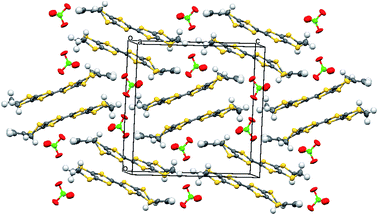 |
| | Fig. 2 Crystal packing of the salt 8·ClO4 viewed down the a axis. | |
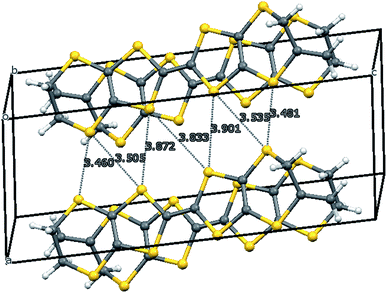 |
| | Fig. 3 Inter-dimer S⋯S contacts (Å) in the a direction in the salt 8·ClO4. | |
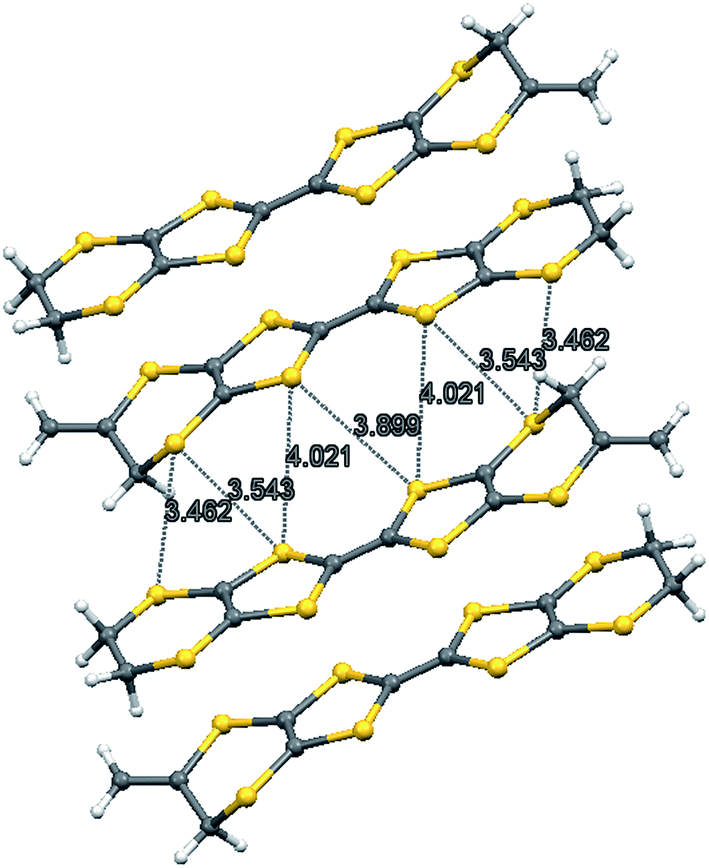
The X-ray crystal structure of the salt formed from donor 8 and iodine showed that it had a composition 2:2:1 donor:I3:I2. Two probe conductivity measurements showed that it was a semiconductor with room temperature conductivity of 1.89 × 10−4 S cm−1 and an energy of activation of 0.373 eV. The crystal system is triclinic and the space group is P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit is composed of one donor cation, one triiodide and half of an iodine molecule, since the iodine molecule lies on a centre of symmetry. The donor cations are organised in face to face pairs, which are almost surrounded by iodine molecules and triiodide anions (Fig. 4 and 5). However, there are side to side contacts between adjacent pairs of donor cations in the a direction (Fig. 6). The I–I bond lengths are 2.8054(13) Å in the iodine molecule and 2.8730(10) and 2.9535(10) Å in the triiodide anion, and the two species approach each other at 85° with a I⋯I contact of 3.3789(13) Å between them. Lines of triiodide anions run through the structure between the donor cation pairs, with successive short and long head to tail I⋯I contacts between triodides of 3.4274(11) and 3.8388(12) Å. Within the radical cation pairs there are four short S⋯S contacts between the S atoms of the TTF core, all of length 3.396(4) Å. In addition there are two sets of two short S⋯S contacts between adjacent cation pairs of length 3.462(4) and 3.543(4) Å (Fig. 6).
. The asymmetric unit is composed of one donor cation, one triiodide and half of an iodine molecule, since the iodine molecule lies on a centre of symmetry. The donor cations are organised in face to face pairs, which are almost surrounded by iodine molecules and triiodide anions (Fig. 4 and 5). However, there are side to side contacts between adjacent pairs of donor cations in the a direction (Fig. 6). The I–I bond lengths are 2.8054(13) Å in the iodine molecule and 2.8730(10) and 2.9535(10) Å in the triiodide anion, and the two species approach each other at 85° with a I⋯I contact of 3.3789(13) Å between them. Lines of triiodide anions run through the structure between the donor cation pairs, with successive short and long head to tail I⋯I contacts between triodides of 3.4274(11) and 3.8388(12) Å. Within the radical cation pairs there are four short S⋯S contacts between the S atoms of the TTF core, all of length 3.396(4) Å. In addition there are two sets of two short S⋯S contacts between adjacent cation pairs of length 3.462(4) and 3.543(4) Å (Fig. 6).
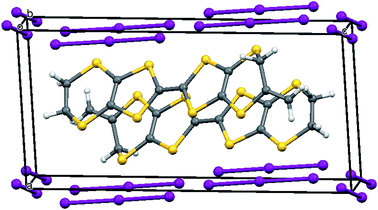 |
| | Fig. 4 A face to face radical cation pair surrounded by triiodide anions and iodine molecules in the crystal structure of 8·I3·0.5I2. | |
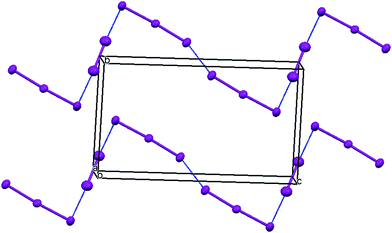 |
| | Fig. 5 Triodide/iodine network in the bc plane in the crystal structure of 8·I3·0.5I2. Intermolecular contacts < 3.5 Å shown. | |
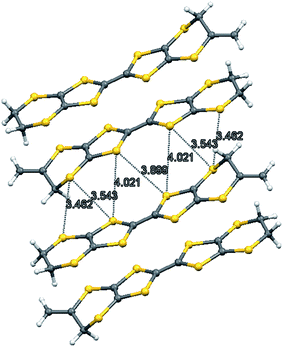 |
| | Fig. 6 S⋯S contacts (Å) between adjacent dimers of donor cations in 8·I3·0.5I2. | |
Synthesis of donors with a side chain alkene group
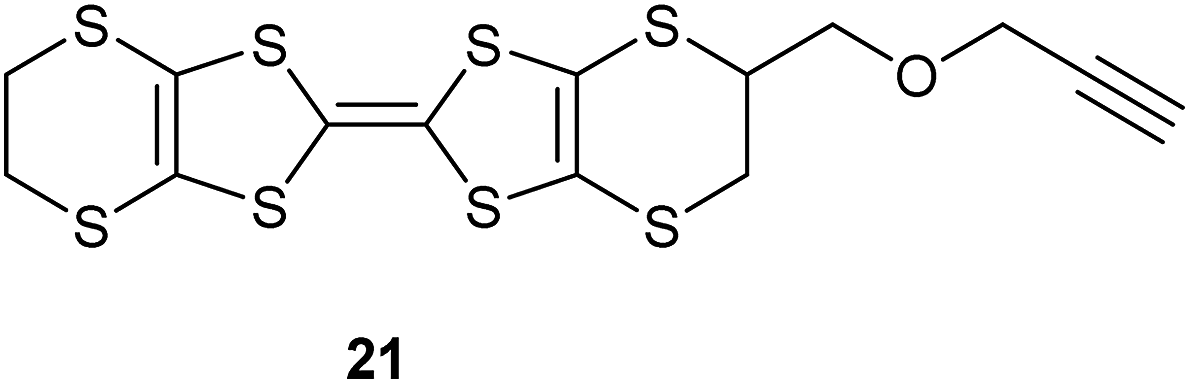
Reaction of diallyl ether with an excess of the trithione 11 in hot toluene gave thione 14, the product of one cycloaddition, as the main product in 68% yield. A small amount of the bis(thione) 15, formed by both alkene groups reacting with a trithione molecule was also isolated in 6% yield. Increasing the amount of the trithione in the reaction did not alter the product distribution. Conversion of the thione 14 to oxo compound 16 with mercuric acetate proceeded in 83% yield, and the oxo compound was coupled with the unsubstituted thione 18 to yield the BEDT-TTF derivative with an allyloxymethyl side chain 9 in 53% yield after separation from homo-coupled products (Scheme 2). The bis(thione) 15 decomposed slowly. Although it can be converted to the corresponding bis(oxo compound) 17, attempts to couple this product with 18 did not lead to any bis(BEDT-TTF) derivatives. The corresponding chemistry starting with diallyl sulphide proceeded in a similar way though with lower yields. Thus, reaction with trithione 11 gave the thione 19 resulting from just one cycloaddition in 31% yield. This was converted in 53% yield to the oxo compound 20, which was then coupled to the unsubstituted thione 18 to give the BEDT-TTF derivative with an allylthiomethyl side chain 10 in 31% yield (Scheme 3). Zhu et al. have reported the closely related alkyne analogue 21.10
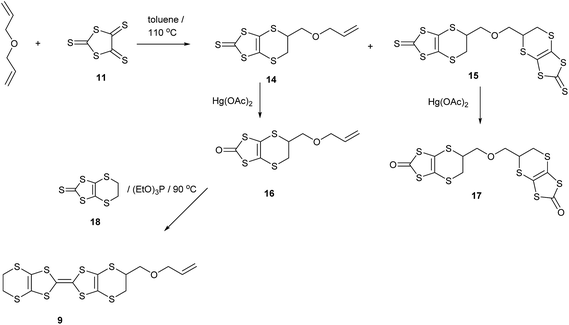 |
| | Scheme 2 | |
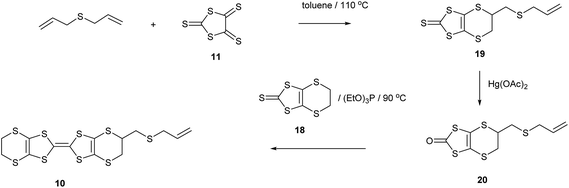 |
| | Scheme 3 | |
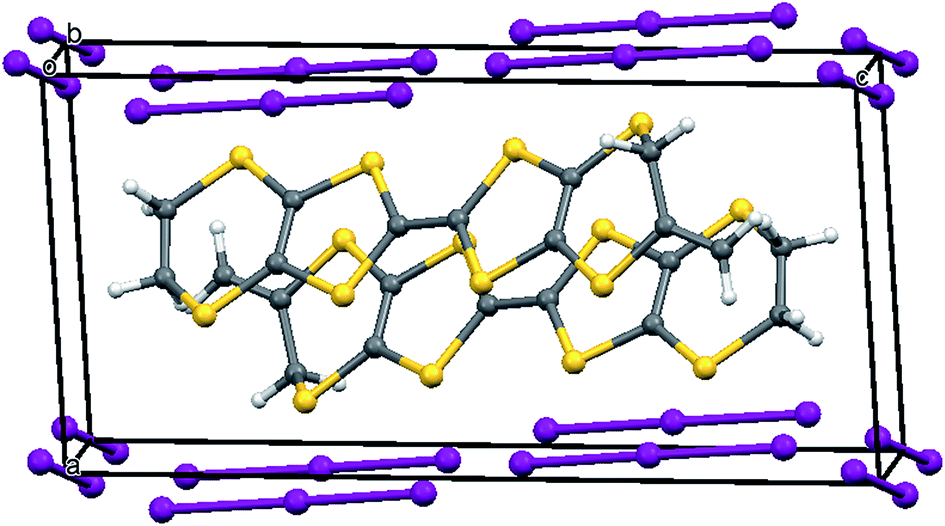
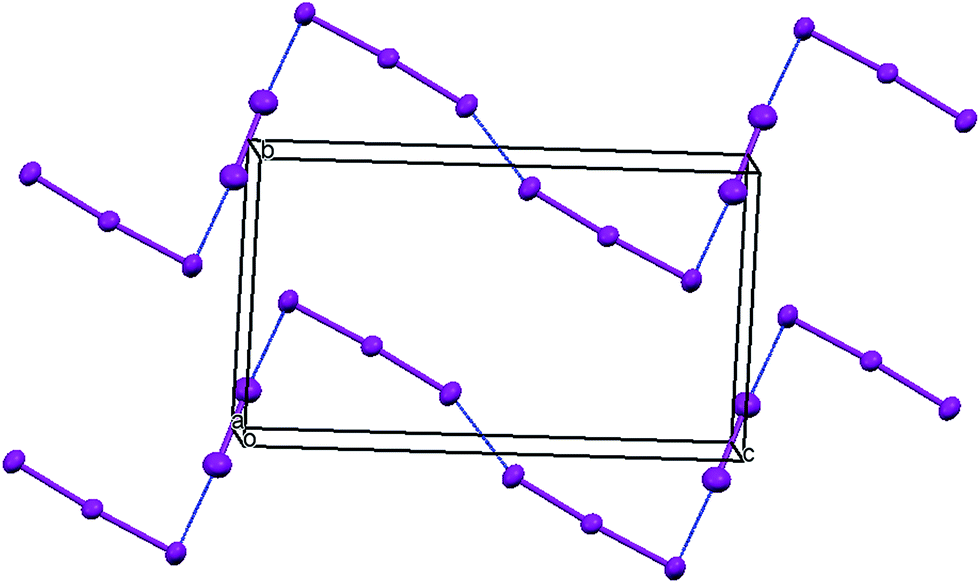
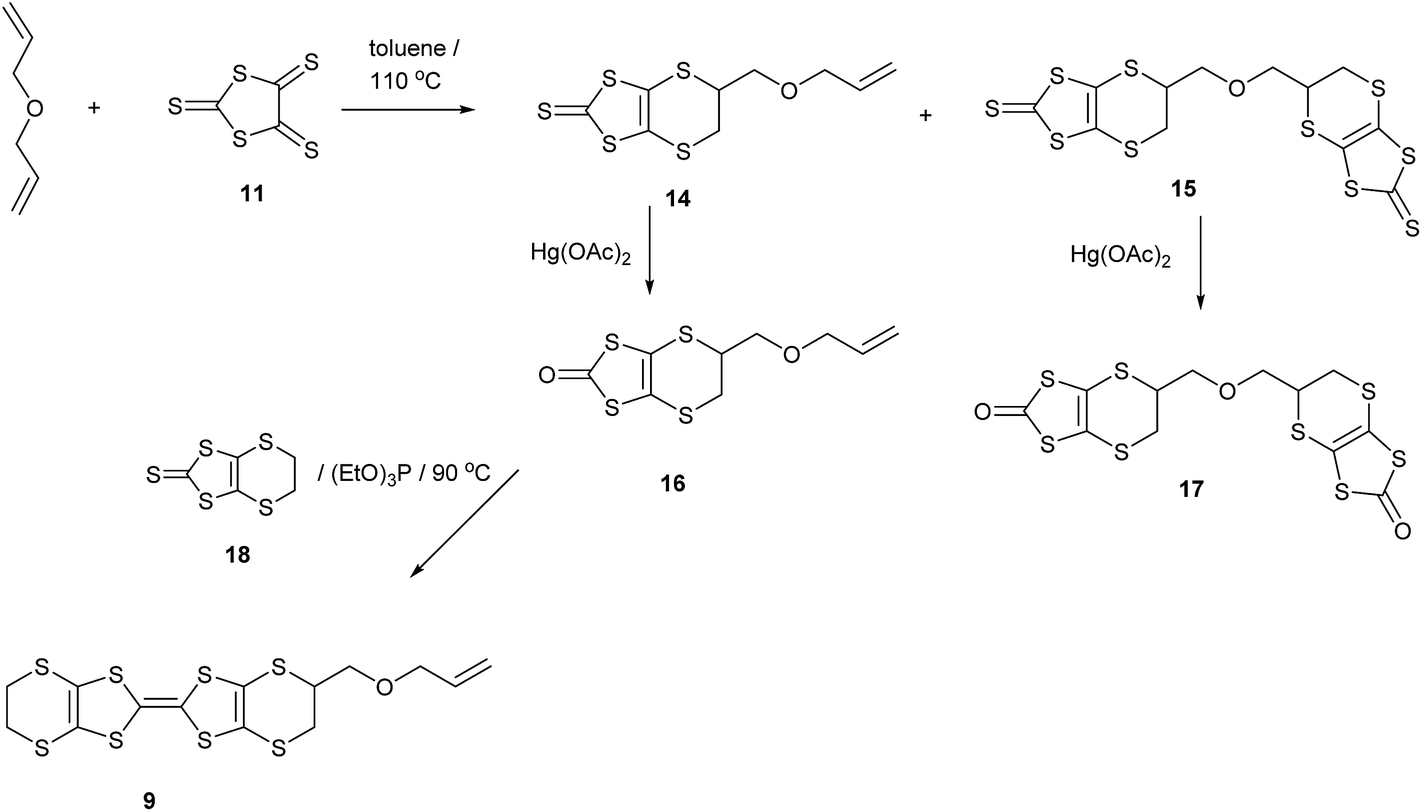
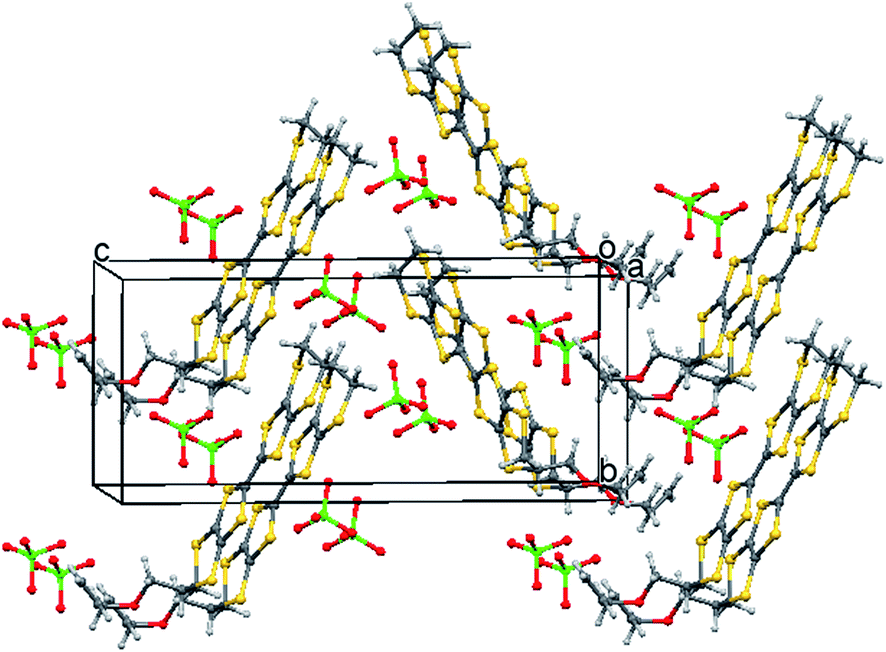
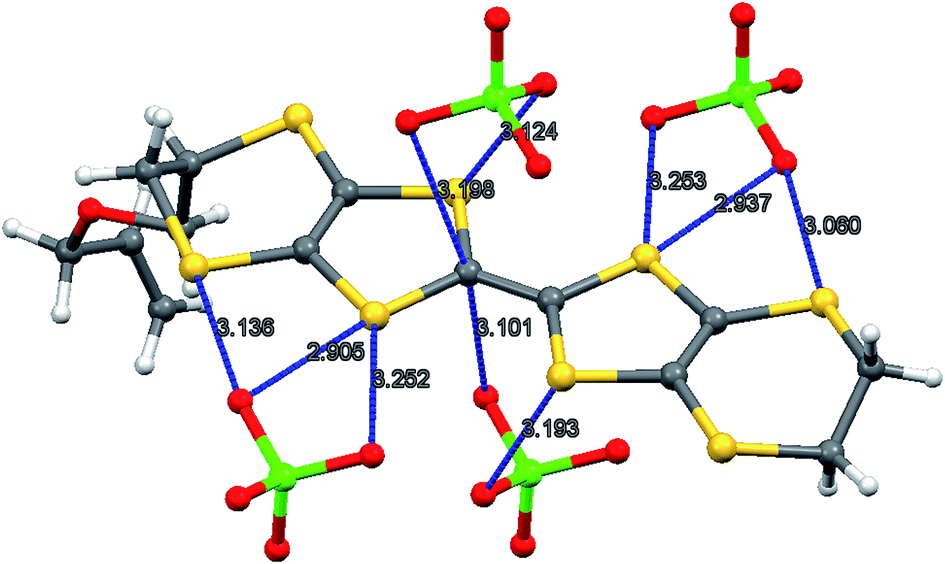
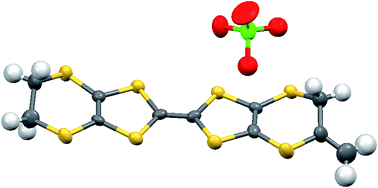
The oxidation potentials of donors 9 and 10 were similar to those of BEDT-TTF. Electrocrystallisation experiments with 9 have yielded a 1:2 (Table 1) perchlorate salt whose crystal structure has been determined. Diffusion experiments with iodine only gave very small crystals of low quality. The crystal structure of 9·2ClO4, measured at 100 K, is monoclinic, in space group P21 and there are two donors each bearing a charge of 2+ and four perchlorate anions in the unit cell. The crystal structure is shown in Fig. 7. The donor dication shows the typically very long bond between dithiole units of 1.44(2) Å, and the four central C–S bonds, shortened by the 2+ charge, lie in the range 1.676(18)–1.717(18) Å similar to those reported for the dication of BEDT-TTF.11 The monosubstituted donor is disordered between two positions, which are roughly related by a 2 fold rotation about the long axis of the molecule. The positions differ in the point of attachment of the side chain to the dithiin ring, however the side chain lies so that the two structures have a common position for the terminal allyloxy group. This is achieved by the dithiin ring in the two structures adopting an approximate envelope conformation with the side chain attached to the “flap” methine carbon and oriented in a pseudo-axial manner (Fig. 8). There is no stacking of donor dications, but there are lines of dications along the a axis organised so that the molecules have edge to edge S⋯S contacts in the range 3.630(7)–4.198(8) Å. Adjacent donors in the b direction are strongly slipped relative to each other so that there are only three S⋯S contacts (3.728(7)–3.785(6) Å) between sulfurs from the unsubstituted end of one donor with sulfurs from the substituted end of the next donor (Fig. 9). The perchlorate anions are distributed among the donor dications, and each donor has S⋯O contacts to four anions (Fig. 10). Although donor stacking and segregated anions are common in BEDT-TTF radical cation salts, the larger donor charge in this case leads to anion contacts with the organosulfur residue. Conductivity measurements showed that the material was an insulator as might be expected.
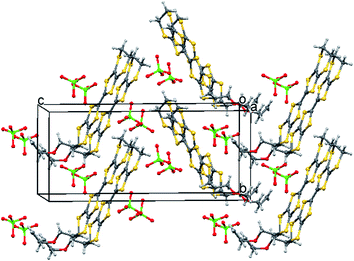 |
| | Fig. 7 Crystal structure of 9·2ClO4 viewed down the a axis, showing the integration of dicationic donors with anions. | |
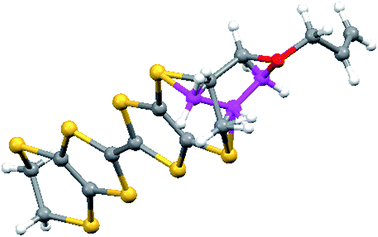 |
| | Fig. 8 View of the donor dication of 9 showing the two disordered orientations of the side chain and how they overlap. | |
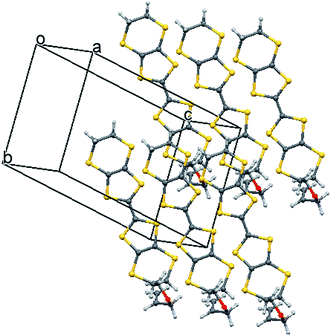 |
| | Fig. 9 Partial crystal packing diagram of 9·2ClO4 showing the relation between donor dications in the a and b directions. Only one conformation of the disordered donor is shown and anions are omitted for clarity. | |
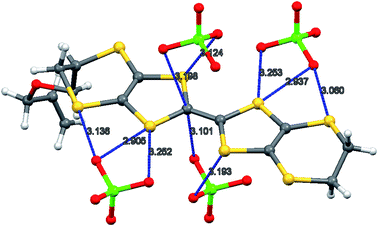 |
| | Fig. 10 Contacts (Å) between the donor dication 92+ and perchlorate anions. | |
Conclusion
The small size of the substituent on exo-methylene-BEDT-TTF 8 provides minimal steric hindrance to crystal packing and may offer access to a wide range of new radical cation salts, as observed for BEDT-TTF. The ready preparation of donors 9 and 10 with a terminal alkene substituent provide donors with the potential for incorporation into polymers. Both of these aspects are now under investigation in our laboratory.
Experimental
General
Solution NMR spectra were measured on a JEOL Eclipse 400 spectrometer at 400 MHz for 1H and at 100.6 MHz for 13C using CDCl3 as solvent and tetramethylsilane (TMS) as standard unless otherwise stated, and measured in p.p.m. downfield from TMS with coupling constants reported in Hz. IR spectra were recorded on a Perkin Elmer Spectrum 100 FT-IR Spectrometer using Attenuated Total Reflection sampling unless otherwise stated, and are reported in cm−1. Mass spectra were recorded at the EPSRC UK National Mass Spectrometry Facility at the University of Swansea. Chemical analysis data were obtained from Mr Stephen Boyer, London Metropolitan University. Flash chromatography was performed on 40–63 silica gel. Cyclic voltammetry was conducted on a Metrohm μ-Autolab Type III.
exo-Methylene-BEDT-TTF, 8
To a solution of tosyloxymethyl-BEDT-TTF 139 (0.103 g, 0.18 mmol) in dry THF (5 ml) under nitrogen was added sodium tert-butoxide (27 mg, 0.28 mmol). The solution was stirred at room temperature for 3 h. The solvent was evaporated, DCM added and the mixture washed with water. The organic phase was collected and dried over Na2SO4. The product was purified by flash chromatography (3:1 hexane:ethyl acetate) to yield donor 8 as an orange solid (0.099 g, 97%), m.p. 165 °C (dec.). δH (400 MHz, CDCl3): 5.22 (1H, s, –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHH), 5.13 (1H, s, –CCHH), 3.52 (2H, s, 6-H2), 3.23 (4H, s, 5′-,6′-H2); δC (100 MHz, CDCl3): 137.4 (CHCH2), 117.1, 114.6, 113.8, 113.7, 113.5, 112.0 (2-,2′-,3a-,3a′-,7a-,7a′-C), 111.0 (–CCH2), 36.5 (6-C), 30.1 (5′,6′-C); νmax: 2958, 2921, 2847, 1598, 1407, 1386, 1282, 1259, 1223, 1211, 1124, 1093, 1020, 899, 875, 799, 769, 729, 622, 530, 496; found C, 33.45; H, 2.09%, C11H8S8 requires C, 33.33; H, 2.08%.
CHH), 5.13 (1H, s, –CCHH), 3.52 (2H, s, 6-H2), 3.23 (4H, s, 5′-,6′-H2); δC (100 MHz, CDCl3): 137.4 (CHCH2), 117.1, 114.6, 113.8, 113.7, 113.5, 112.0 (2-,2′-,3a-,3a′-,7a-,7a′-C), 111.0 (–CCH2), 36.5 (6-C), 30.1 (5′,6′-C); νmax: 2958, 2921, 2847, 1598, 1407, 1386, 1282, 1259, 1223, 1211, 1124, 1093, 1020, 899, 875, 799, 769, 729, 622, 530, 496; found C, 33.45; H, 2.09%, C11H8S8 requires C, 33.33; H, 2.08%.
Radical cation salts of exo-methylene-BEDT-TTF, 8
Perchlorate salt.
To the anodic side of a H-shaped electrochemical cell fitted with a glass frit was placed donor 8 (5 mg, 0.0126 mmol), and to the cathodic side of the cell was added a 0.0089 M solution of tetrabutylammonium perchlorate in chlorobenzene (40 ml). The level of solution in each compartment was allowed to equilibrate, and into each side was inserted a platinum-tipped electrode. A constant current of 2.0 μA was applied across the cell for 3 days to give black needle-shaped crystals of 8·ClO4.
Triiodide salt.
A solution of donor 8 (10 mg) in dichloromethane (10 ml) was slowly diffused into a solution of iodine (10 mg) in benzonitrile (10 ml) over two weeks to give black crystals of 8·I3·0.5I2.
Syntheses of alkenyl-substituted donors 9 and 10
5-(Allyloxymethyl)-5,6-dihydro-1,3-dithiolo[4,5-b]1,4-dithiin-2-thione, 14.
A suspension of trithione 11 (2.94 g, 15.0 mmol) and diallyl ether (0.49 g, 5.0 mmol) in toluene (50 ml) was heated to reflux overnight, cooled to RT and filtered. The solid residue was washed with toluene and the combined filtrates collected. The solvent was evaporated and the residue purified by flash chromatography on silica (6:1 = cyclohexane:ethyl acetate) (Rf 0.75) to yield compound 14 (0.99 g, 68%) as a red oil. δH (400 MHz, CDCl3): 5.81–5.91 (1H, m, –CHCH2), 5.27 (1H, dq, J = 17.3, 1.6 Hz, –CHCHH-trans), 5.20 (1H, dq, J = 10.1, 1.6 Hz, –CHCHH-cis), 4.01 (2H dt, J = 5.7, 1.2 Hz, –O–CH2–CHCH2), 3.83–3.89 (1H, m, 5 H), 3.77 (1H, dd, J = 9.5, 8.5 Hz, 5-CHα), 3.60 (1H, dd, J = 9.5, 5.5 Hz, 5-CHβ), 3.27–3.36 (2H, m, 6-H2); δC (100 MHz, CDCl3): 207.7 (CS), 133.7 (–CHCH2), 123.5, 122.1 (3a-,7a-C), 117.8 (–CHCH2), 72.3 (–O–CH2–CHCH2), 70.6 (5-CH2–O–), 42.6 (5-C), 31.3 (6-C); νmax: 2928, 2852, 1644, 1469, 1448, 1408, 1346, 1283, 1262, 1093, 1061, 989, 909, 770, 679; HRMS: (ASAP) found: 294.9403 (100%), C9H10O1S5 +H requires: 294.9408; found C, 36.86; H, 3.46%, C9H10O1S5 requires C, 36.73; H, 3.40%.
Further elution of the column gave bis(2-thioxo-1,3-dithiolo[4,5-b]1,4-dithiin-5-methyl)ether 15 (Rf 0.35) (0.15 g, 6%) as a brown solid, m.p. 117 °C (dec). δH (400 MHz, CDCl3): 3.84–3.93 (4H, m, –CH2–O–CH2), 3.69–3.77 (2H, m, 5-,5′-H), 3.29–3.34 (4H, m, 6-,6′-H2); δC (100 MHz, CDCl3): 207.5 (2 × CS), 122.8, 121.9 (3a-,3a′-,7a-,7a′-C), 76.7 (–CH2–O–CH2–). 42.1 (5-,5′-C), 31.2 (6-,6′-C); νmax: 2847, 1478, 1404, 1261, 1112, 1054, 868, 517; HRMS: (ASAP) found: 490.8003, C12H10O1S10 + H requires 490.8011; found C, 29.48; H, 2.15%, C12H10OS10 requires C, 29.38; H, 2.04%. Over a period of time this compound decomposed.
5-(Allyloxymethyl)-5,6-dihydro-1,3-dithiolo[4,5-b]1,4-dithiin-2-one, 16.
To a solution of thione 14 (2.13 g, 7.3 mmol) in CHCl3 (100 ml) was added mercury(II) acetate (5.70 g, 17.9 mmol) and the suspension left stirring for 4 h. under nitrogen at RT. The solid obtained during the reaction was filtered and washed with CHCl3. The combined filtrates were neutralised with solid NaHCO3. The organic layer was collected and washed with water, dried over MgSO4 and the solvent evaporated to give the desired compound 16 (2.70 g, 83%) as a yellow-brown oil; δH (400 MHz, CDCl3): 5.77–5.87 (1H, m, –CHCH2), 5.23 (1H, dq, J = 17.2, 1.4 Hz, –CHCHH-trans), 5.16 (1H, dq, J = 10.4, 1.4 Hz, –CHCHH-cis), 3.97 (2H, dt, J = 5.7, 1.3 Hz, –O–CH2–CHCH2), 3.82–3.88 (1H, m, 5-H), 3.75 (1H, t, J = 9.6 Hz, 5-CHα), 3.28 (1H, dd, J = 9.6, 5.6 Hz, 5-CHβ), 3.23–3.32 (2H, m, 6-H2); δC (100 MHz, CDCl3): 188.7 (CO), 133.8 (–CHCH2), 113.9, 112.5 (3a-,7a-C), 117.7 (CHCH2), 72.3 (O–CH2–CHCH2), 70.7 (5-CH2–O–), 44.3 (6-C), 32.4 (5-C); νmax: 2918, 2853, 1673, 1621, 1509, 1241, 1093, 825, 763, 542; HRMS: (ASAP) found: 278.9632, C9H10O2S4 + H requires: 278.9636; found C, 38.69; H, 3.48%, C9H10O2S4 requires C, 38.85; H, 3.60%.
(Allyloxymethyl)-BEDT-TTF, 9.
To a solution of oxo compound 16 (1.08 g, 3.90 mmol) in freshly distilled triethyl phosphite (30 ml) was added unsubstituted thione 18 (1.74 g, 7.80 mmol) and the solution was left stirring for 5 h. under nitrogen at 90 °C. The BEDT-TTF formed was filtered off and the triethyl phosphite was distilled off from the filtrate using a Kugelrohr apparatus. The residue was purified using flash chromatography (3:1 = cyclohexane:ethyl acetate) affording the desired product 9 as an orange solid (0.94 g, 53%), which was recrystallized from acetonitrile, m.p. 82–84 °C. δH (400 MHz, CDCl3): 5.83–5.94 (1H, m, –CHCH2), 5.29 (1H, dq, J = 16.9, 1.8 Hz, –CHCHHtrans), 5.22 (1H, dd, J = 10.5, 1.8 Hz, –CH = CHHcis), 4.02 (2H, dt, J = 5.5, 1.2 Hz, –O–CH2–CHCH2), 3.78–3.84 (1H, m, 5-H), 3.74 (1H, t, J = 9.2 Hz, 5-CHα), 3.57 (1H, dd, J = 9.2, 5.0 Hz, 5-CHβ), 3.29 (4H, s, 5′-,6′-H2), 3.19–3.28 (2H, m, 6-H2); δC (100 MHz, CDCl3): 134.0 (–CHCH2), 117.7 (CHCH2), 114.2, 113.7, 113.3, 111.8, 111.6 (sp2-C), 72.3 (O–CH2–CHCH2), 71.0 (5-CH2), 43.0 (5-C), 32.1 (6-C), 30.1 (5′-, 6′-C); νmax: 3075, 2977, 2929, 2847, 1644, 1472, 1448, 1408, 1347, 1283, 1260, 1094, 986, 907, 770, 680, 652, 555, 500, 449; HRMS: (ASAP) found: 454.8881, C14H14OS8 + H requires: 454.8883; found C, 36.91; H, 3.15%, C14H14OS8 requires C, 37.00; H, 3.08%.
Bis(2-oxo-1,3-dithiolo[4,5-b]1,4-dithiin-5-methyl)ether, 17.
To a solution of 15 (0.75 g, 1.53 mmol) in CHCl3 (60 ml) was added mercury(II) acetate (1.71 g, 5.36 mmol) and the suspension was left stirring at RT for 5 h. The solid was filtered off and the solution neutralised with saturated aqueous NaHCO3 (5 × 50 ml). The organic phase was collected, washed sequentially with water (50 ml) and brine (50 ml) and dried over MgSO4. Evaporation of the solvent gave the desired product 17 as a brown solid (0.64 g, 91%), m.p. 122–124 °C. δH (400 MHz, DMSO-d6): 4.14–4.21 (2H, m, 5-, 5′-H), 3.83–3.91 (2H, m) and 3.67–3.73 (2H, m) (–CH2–O–CH2–), 3.48 (2H, dd, J = 13.6, 3.4 Hz) and 3.39 (2H, dd, J = 13.6, 6.3 Hz) (6-,6′-H2); δC (100 MHz, DMSO-d6): 187.9 (2 × CO), 114.2, 112.7 (3a-,3a′-,7a-,7a′-C), 71.4 (CH2–O–CH2–), 44.4 (5-, 5′-C), 32.0 (6-, 6′-C); νmax: 2850, 1614, 1498, 1408, 1100, 768, 463; HRMS: (ASAP) found 458.8489, C12H10O3S8 + H requires 458.8468; found C, 31.54; H, 2.29%, C12H10O3S8 requires C, 31.44; H, 2.18%. Attempted cross coupling of 17 with thione 18 in triethyl phosphite was unsuccessful.
5-(Allylthiomethyl)-5,6-dihydro-1,3-dithiolo[4,5-b]1,4-dithiin-2-thione, 19.
A suspension of trithione 11 (5.82 g, 29.7 mmol) and diallyl sulphide (1.42 g, 11.9 mmol) in toluene (160 ml) was heated to reflux overnight, cooled to RT and filtered. The solid residue was washed with CHCl3 and the combined filtrates collected. The solvent was evaporated and the residue purified by flash chromatography on silica (3:1 = cyclohexane:ethyl acetate) to yield compound 19 (1.14 g, 31%) as a red oil. δH (400 MHz, CDCl3): 5.68–5.78 (1H, m, –CHCH2), 5.10 (1H, br d, J = 9.6 Hz, –CHCHHcis), 5.09 (1H, dd, J = 17.1, 1.4 Hz, –CHCHHtrans), 3.66–3.72 (1H, m, 5-H), 3.30–3.41 (2H, m, 6-H2), 3.14 (2H, d, J = 7.2 Hz, CH2–CHCH2), 2.87 (1H, dd, J = 14.0, 8.8 Hz, 5-CHα), 2.80 (1H, dd, J = 14.0, 5.9 Hz, 5-CHβ); δC (100 MHz, CDCl3): 207.8 (CS), 133.7 (–CHCH2), 123.1, 121.7 (3a-,7a-C), 118.2 (–CHCH2), 42.5 (5-C), 35.3 (5-CH2-S), 34.8 (–S–CH2–CHCH2), 32.9 (6-C); νmax: 2905, 1631, 1484, 1403, 1054, 917, 574; HRMS: (ASAP) found: 310.9179, C9H11S6 + H requires: 310.9180.
5-(Allylthiomethyl)-5,6-dihydro-1,3-dithiolo[4,5-b]1,4-dithiin-2-one, 20.
To a solution of thione 19 (1.10 g, 3.5 mmol) in CHCl3 (25 ml) under nitrogen was added mercury(II) acetate (3.00 g, 9.4 mmol) and the suspension was left stirring for 2 h. at RT. The solid obtained during the reaction was filtered and washed with CHCl3. The combined filtrates were washed with a saturated solution of NaHCO3 (5 × 20 ml). The organic layer was collected and washed sequentially with water and brine and dried over MgSO4 to give the desired compound 20 (0.60 g, 53%) as a pink-red oil. δH (400 MHz, CDCl3): 5.67–5.78 (1H, m, –CHCH2), 5.10 (1H, br d, J = 10.0 Hz, –CHCHHcis), 5.09 (1H, dd, J = 17.5, 1.3 Hz, –CHCHHtrans), 3.69–3.75 (1H, m, 5-H), 3.39 (1H, dd, J = 13.4, 3.1 Hz, 6-Hα), 3.33 (1H, dd, J = 13.4, 6.0 Hz, 6-Hβ), 3.14 (2H, d, J = 7.3 Hz, –CH2–CHCH2), 2.89 (1H, dd, J = 13.9, 9.0 Hz, 5-CHα), 2.81 (1H, dd, J = 13.9, 5.8 Hz, 5-CHβ); δC (100 MHz, CDCl3): 188.9 (CO), 133.8 (–CHCH2), 118.2 (CHCH2), 113.5, 112.3 (3a-,7a-C), 44.4 (5-C), 35.4 (5-CH2–S), 34.9 (–S–CH2–CHCH2), 34.2 (6-C); νmax: 2910, 1670, 1628, 1503, 1405, 1226, 988, 916, 888, 754; found C, 36.60; H, 3.51%, C9H10OS5 requires C, 36.73; H, 3.40%.
(Allylthiomethyl)-BEDT-TTF, 10.
To a solution of oxo compound 20 (0.60 g, 2.05 mmol) in freshly distilled triethyl phosphite (30 ml) was added unsubstituted thione 18 (0.92 g, 4.1 mmol) and the solution was left stirring for 5 h. under nitrogen at 90 °C. The BEDT-TTF formed during the reaction was filtered off and the triethyl phosphite was distilled off from the filtrate using a Kugelrohr apparatus. The crude red oil was purified by flash chromatography (6:1 = cyclohexane:ethyl acetate) affording the donor 10 as an orange-red solid (0.30 g, 31%), m.p. 107–109 °C. δH (400 MHz, CDCl3): 5.67–5.77 (1H, m, –CHCH2), 5.08 (1H, br d, J = 10.8 Hz, –CHCHHcis), 5.07 (1H, dq, J = 16.4, 1.2 Hz, –CHCHHtrans), 3.58–3.63 (1H, m, 5-H), 3.21–3.27 (2H, m, 6-H2), 3.22 (4H, s, 5′-,6′-H2), 3.12 (2H, d, J = 7.2 Hz, –S–CH2CHCH2), 2.82 (1H, dd, J = 14.0, 9.2 Hz, 5-CHα) 2.75 (1H, dd, J = 14.0, 5.8 Hz, 5-CHβ); δC (100 MHz, CDCl3): 133.7 (–CHCH2), 118.0 (–CHCH2), 113.8, 113.7, 112.8, 111.9, 111.6 (2-,2′-,3a-,3a′-,7a-,7a′-C), 42.9 (5-C), 35.3 (–S–CH2–CHCH2), 34.9 (5-CH2S–), 33.6 (6-C), 30.1 (5′-, 6′-C); νmax: 2915, 1633, 1517, 1399, 1287, 1203, 913, 882, 772; found C, 35.81; H, 2.93%, C14H14S9 requires C, 35.74; H, 2.98%.
Radical cation salts of allyloxymethyl-BEDT-TTF, 9
Perchlorate salt.
To the anodic side of a H-shaped electrochemical cell fitted with a glass frit was placed donor 9 (10 mg, 0.022 mmol), and to the cathodic side of the cell was added a 0.0089 M solution of tetrabutylammonium perchlorate in chlorobenzene (40 mL). The level of solution in each compartment was allowed to equilibrate, and into each side was inserted a platinum-tipped electrode. A constant current of 0.5 μA was applied across the cell for 4 weeks to give black needle shaped crystals of 9·(ClO4)2.
X-ray crystallography
Crystal data for 8·ClO4.
C11H8S8·ClO4, Mr = 496.10, monoclinic, a = 6.401(2), b = 16.234(5), c = 16.417(5) Å, β = 95.579(9)°, V = 1697.9(9) Å3, Z = 4, P21/c, Dc = 1.941 g cm−3, MoKα (λ = 0.71073 Å), μ = 1.224 mm−1, T = 100(2) K, 12665 unique reflections (Rint = 0.16), 8291 with F2 > 2σ, R(F, F2 > 2σ) = 0.11, Rw (F2, all data) = 0.16. The data were measured at the National Crystallography Centre,12 Southampton University on a Rigaku AFC12 diffractometer equipped with enhanced sensitivity (HG) Saturn724+ CCD detector mounted at the window of an FR-E+ SuperBright molybdenum rotating anode generator (Mo Kα, λ = 0.71075 Å) with VHF Varimax optics (70 μm focus) using Crystal Clear software13 for data collection and reduction. The structure was solved with SHELXS97,14 and refined with SHELX201315 within OLEX2.16 Data were collected from a twinned crystal. ROTAX17 was used to identify the twin law, corresponding to a 2-fold rotation about the 1 0 0 direction: twin law [1.000 0.000 0.000] [0.000 −1.000 0.000] [−0.498 0.000 −1.000] and the twin component fraction refined to 0.71. Disorder in the ClO4 anion was modelled with two of the oxygen sites split over two sites with the occupancy refined competitively, 0.63/0.37(3), and isotropic atomic displacement parameters for partially occupied atoms. Distance restraints were applied to the Cl–O distances.
Crystal data for 8·I3·(I2)0.5.
C11H8S8·I3·0.5I2, Mr = 904.28, triclinic, a = 8.2014(15), b = 9.1986(16), c = 15.637(3) Å, α = 86.967(10), β = 86.049(10), γ = 72.972(7)°, V = 1124.6(4) Å3, Z = 2, P, Dc = 2.670 g cm−3, MoKα (λ = 0.71073 Å), μ = 6.281 mm−1, T = 296(2) K, 4911 unique reflections (Rint = 0.026), 3208 with F2 > 2σ, R(F, F2 > 2σ) = 0.049, Rw (F2, all data) = 0.15. Data were collected at room temperature using a Rigaku Mercury sII CCD configured with the Rigaku MicroMax-007HF generator and VariMax confocal mirror using Crystal Clear software13 for data collection and reduction. The structure was solved by SIR9218 and refined with SHELX201315 using the Rigaku CrystalStructure® software package.
Crystal data for 9·(ClO4)2.
C14H14OS8·2ClO4, Mr = 653.63, monoclinic, a = 5.9156(4), b = 9.5177(7), c = 21.3112(16) Å, β = 97.871(4)°, V = 1188.58(13) Å3, Z = 2, P21, Dc = 1.826 g cm−3, MoKα (λ = 0.6889 Å), μ = 0.942 mm−1, T = 100(2) K, 9058 unique reflections (Rint = 0.08), 8296 with F2 > 2σ, R(F, F2 > 2σ) = 0.079, Rw (F2, all data) = 0.22. The data were collected using synchrotron X-ray radiation (λ = 0.6889 Å) at the Diamond Light Source, UK using Beamline I19 situated on an undulator insertion device with a combination of double crystal monochromator, vertical and horizontal focussing mirrors and a series of beam slits (primary white beam and either side of the focussing mirrors). The experimental hutch (EH1) is equipped with a Crystal Logic 4-circle kappa geometry goniometer with a Rigaku Saturn 724 CCD detector and an Oxford Cryosystems Cryostream plus cryostat (80–500 K). Data reduction, cell refinement and absorption correction were performed using CrystalClear-SM Expert 2.0 r5.13 The structure was solved with SUPERFLIP19 and refined with SHELXL201315 within OLEX2 Graphics in OLEX2.16 The structure was refined as a 2-component twin; twin law (1 0 0/0 −1 0/−1 0 −1) corresponding to 2-fold rotation about the 1 0 0 direction. Twin law identified using ROTAX and hklf5 file generated through WINGX.20 Twin scale factor refined to 0.394(4). Disorder was present in the sp3 carbon atoms of the substituted dithiin ring and the first methylene carbon of the side chain, and these were modelled over two half occupied sites. Distance restraints were applied and atoms were refined with isotropic displacement parameters. All geometric analyses were performed using PLATON21 and all illustrations were made with MERCURY.22 Data is deposited at the Cambridge Crystallographic Data Centre with code numbers CCDC 1051494–1051496.
Conductivity Measurements
Two-probe DC transport measurements were made on several crystals of 8·I3·0.5I2 using a HUSO HECS 994C multi-channel conductometer, and on crystals of 9·(ClO4)2 using an Oxford Instruments Optistat and temperature controller. Gold wires were attached to the crystal, and the attached wires were connected to an four-pin integrated circuit plug with conductive cement.
Acknowledgements
We thank Nottingham Trent University for PhD grants (MZ, JRL) and the EPSRC UK National Mass Spectrometry Facility at Swansea University for data. We thank EPSRC for funding the National Crystallography Service including access to Synchrotron facilities at the Diamond Light Source, LM thanks the Royal Society for Research Grants (RG100853 and RG081209), an International Exchange Scheme (IE130367) and an International Joint Project (JP0869972). LM thanks the Royal Society of Chemistry for a Journals Grant for International Authors.
References
-
TTF Chemistry: Fundamentals and Applications of Tetrathiafulvalene, ed. J.-I. Yamada and T. Sugimoto, Springer, 2004 CrossRef CAS; A. Ardavan, S. Brown, S. Kagoshima, K. Kanoda, K. Kuroki, H. Mori, M. Ogata, S. Uji and J. Wosnitza, J. Phys. Soc. Jpn., 2012, 81, 011004 CrossRef CAS; N. Avarvari and J. D. Wallis, J. Mater. Chem., 2009, 19, 4061–4076 RSC; C. Rovira, Chem. Rev., 2004, 104, 5289–5317 CrossRef PubMed; M. Herranz, L. Sanchez and N. Martin, Phosphorus, Sulfur Silicon Relat. Elem., 2005, 180, 1133–1148 CrossRef.
- H. Jiang, X. Yang, Z. Cui, Y. Liu, H. Li, W. Hu and C. Kloc, CrystEngComm, 2014, 16, 5968–5983 RSC; D. Canevet, M. Salle, G. Zhang, D. Zhang and D. Zhu, Chem. Commun., 2009, 2245–2269 RSC; J.-P. Griffiths and J. D. Wallis, J. Mater. Chem., 2005, 15, 347–365 RSC; M. R. Bryce, J. Mater. Chem., 2000, 10, 589–598 RSC.
- H. Kobayashi, A. Kobayashi, T. Nakamura, T. Nogami and Y. Shirota, Chem. Lett., 1987, 559–562 CrossRef CAS.
- S. Iwasaka, T. Nogami and Y. Shirota, Synth. Met., 1988, 26, 177–184 CrossRef CAS; T. Nakamura, S. Iwasaka, H. Nakano, K. Inoue, T. Nogami and H. Mikawa, Bull. Chem. Soc. Jpn., 1987, 60, 365–368 CrossRef; T. Nogami, K. Inoue, T. Nakamura, S. Iwasaka, H. Nakano and H. Mikawa, Synth. Met., 1987, 19, 539–544 CrossRef.
- A. C. Brooks, P. Day, S. I. G. Dias, S. Rabaca, I. C. Santos, R. T. Henriques, J. D. Wallis and M. Almeida, Eur. J. Inorg. Chem., 2009, 3084–3093 CrossRef CAS; E. Ertas and T. Ozturk, Chem. Commun., 2000, 2039–2040 RSC; K. Inoue, Y. Tasaka, O. Yamazaki, T. Nogami and H. Mikawa, Chem. Lett., 1986, 15, 781–784 CrossRef.
- H. Nakano, K. Miyawaki, T. Nogami, Y. Shirota, S. Harada and N. Kasai, Bull. Chem. Soc. Jpn., 1989, 62, 2604–2607 CrossRef CAS.
- H. Nakano, K. Miyawaki, T. Nogami, Y. Shirota, S. Harada, N. Kasai, A. Kobayashi, R. Kato and H. Kobayashi, Bull. Chem. Soc. Jpn., 1990, 63, 2281–2287 CrossRef CAS; V. E. Korotkov, R. P. Shibaeva, K. A. Balodis, V. Y. Khodorkovskii and O. Y. Neiland, Kristallografiya, 1991, 36, 113–116 Search PubMed; A. D. Dubrovskii, N. G. Spitsina, A. N. Chekhlov, O. A. Dyachenko, L. I. Buravov, A. A. Lobach, J. V. Gancedo and C. Rovira, Synth. Met., 2004, 140, 171–176 CrossRef.
- F. Turksoy, J. D. Wallis, U. Tunca and T. Ozturk, Tetrahedron, 2003, 59, 8107–8116 CrossRef CAS.
- Q. Wang, P. Day, J. P. Griffiths, H. Nie and J. D. Wallis, New J. Chem., 2006, 1790–1800 RSC.
- W. Qin and D. Zhu, Gaofenzi
Xuebao, 1997, 121–124 CAS.
- L.-K. Chou, M. A. Quijada, M. B. Clevenger, G. F. de Oliveira, K. A. Abboud, D. B. Tanner and D. R. Talham, Chem. Mater., 1995, 7, 530–534 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683–689 RSC.
-
Crystal Clear-SM Expert software, Rigaku, 2013 Search PubMed.
- G. M. Sheldrick, SHELXS97, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
-
G. M. Sheldrick, SHELX2013, University of Göttingen, Germany, 2012 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- S. Parsons, R. O. Gould, R. I. Cooper and D. J. Watkin, ROTAX, J. Appl. Crystallogr., 2002, 35, 168–174 Search PubMed.
- A. Altomare, M. C. Burla, G. Camalli, G. Cascarano, C. Giacovazzo, A. Gualiardi and G. Polidori, SIR92, J. Appl. Crystallogr., 1994, 27, 435 Search PubMed.
- L. Palatinus and G. Chapuis, SUPERFLIP, J. Appl. Crystallogr., 2007, 40, 786–790 CrossRef CAS.
- L. J. Farrugia, WINGX, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- A. L. Spek, PLATON, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 148–155 CrossRef CAS PubMed.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, MERCURY, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a