
Production of ultra-high concentration calcium alginate beads with prolonged dissolution profile†
Wan-Ping Vooa,
Boon-Beng Leeb,
Ani Idris c,
Aminul Islamde,
Beng-Ti Teyaf and
Eng-Seng Chan*af
c,
Aminul Islamde,
Beng-Ti Teyaf and
Eng-Seng Chan*af
aChemical Engineering Discipline, School of Engineering, Monash University Malaysia, Jalan Lagoon Selatan, 46150 Bandar Sunway, Selangor, Malaysia. E-mail: chan.eng.seng@monash.edu; Fax: +603 55146207; Tel: +603 55145821
bSchool of Bioprocess Engineering, Universiti Malaysia Perlis, 02600 Arau, Perlis, Malaysia
cDepartment of Bioprocess Engineering, Faculty of Chemical and Natural Resource Engineering, Universiti Teknologi Malaysia, 81310 Skudai, Johor, Malaysia
dCatalysis and Science Research Center, Faculty of Science, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
eDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 Serdang, Selangor, Malaysia
fMultidisciplinary Platform of Advanced Engineering, Monash University Malaysia, Jalan Lagoon Selatan, 46150 Bandar Sunway, Selangor, Malaysia
First published on 14th April 2015
Abstract
Calcium alginate hydrogel beads have been widely studied as a carrier matrix for the delivery of food and pharmaceutical compounds. Typically, calcium alginate beads have a short dissolution time of between 1 to 2 h in simulated intestinal fluid, thus limiting some applications that require prolonged release of compounds. This study was aimed at fabricating calcium alginate beads with a prolonged dissolution profile by increasing the alginate concentration beyond the critical viscosity limit (approximately 5000 mPa s) that can be processed using the existing extrusion-dripping system. A temperature-controlled extrusion method was developed and the feasibility of producing alginate beads at ultra-high concentration (UHC) with an initial viscosity ranging from 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mPa s to 353000 mPa s was studied for the first time. The operating temperatures studied ranged from 40 °C to 80 °C. Spherical UHC alginate beads were successfully produced using an alginate solution with an initial viscosity of 33000 mPa s. Fourier-transform infrared (FTIR) analysis indicated that the chemical properties of the alginate gels were not affected by the operating temperature. The dissolution time of the UHC alginate beads was three times longer than that of the beads produced using the normal alginate concentration. In addition, the UHC alginate beads have a unique internal structure that differs from the normal beads. In conclusion, a facile method to produce the UHC alginate beads, without the need for chemical modification of the beads or/and additional processing steps, is demonstrated. The long dissolution time of the UHC alginate beads opens up windows of opportunity for applications in the sustained delivery of drugs and food ingredients.
000 mPa s to 353000 mPa s was studied for the first time. The operating temperatures studied ranged from 40 °C to 80 °C. Spherical UHC alginate beads were successfully produced using an alginate solution with an initial viscosity of 33000 mPa s. Fourier-transform infrared (FTIR) analysis indicated that the chemical properties of the alginate gels were not affected by the operating temperature. The dissolution time of the UHC alginate beads was three times longer than that of the beads produced using the normal alginate concentration. In addition, the UHC alginate beads have a unique internal structure that differs from the normal beads. In conclusion, a facile method to produce the UHC alginate beads, without the need for chemical modification of the beads or/and additional processing steps, is demonstrated. The long dissolution time of the UHC alginate beads opens up windows of opportunity for applications in the sustained delivery of drugs and food ingredients.
1 Introduction
Alginate, which consists of β-D-mannuronic acid (M) and α-L-guluronic acid (G) residues, is a naturally occurring biopolymer found in great abundance in brown seaweeds. The two uronic acids occur in homopolymeric M-blocks, G-blocks, and heteropolymeric random MG-blocks.1 Alginate forms a semi-permeable hydrogel under mild conditions at room temperature following the addition of divalent cations, such as Ca2+. The counter ions will bind to the carboxylates in the G-blocks to form ‘egg-box’ structures.2Alginate hydrogels are biodegradable, biocompatible, and non-toxic, which allows them to be used as drug delivery systems for controlled release of both hydrophobic and hydrophilic drugs.4–6 However, alginate hydrogels have a very short dissolution time in the simulated intestinal fluid (i.e., approximately 1–2 h).7–9 Thus, alginate beads are limited to short term release applications. Alginate hydrogels with prolonged dissolution time may be desirable for applications which require prolonged release of drugs, such as pain relievers, anti-depressants or attention deficit hyperactivity disorder medication for children.10 Alginate hydrogel with prolonged dissolution time are beneficial in sustaining the drug concentration in blood.
Many studies have been done to extend the dissolution time of alginate hydrogel beads by modifying the beads properties. For example, coating the alginate beads with other materials, such as chitosan, Eudragit and whey protein,3,11,12 blending the alginate with other materials, such as pectin and carboxymethyl cellulose,3,13 covalently crosslinking alginate with other materials, such as poly(ethylene glycol)-diamines and polyacrylamide14,15 and chemically modifying alginate to become hydrophobic.16,17
However, these approaches require chemical modification of the beads and/or additional processing step, which may increase the processing costs and give rise to safety and biocompatibility issues. In this respect, a delivery system based on naturally unmodified alginate polymer is still favored since alginate is known to be safe for in vivo applications. One strategy to prolong the dissolution time of alginate beads is by increasing the concentration of alginate polymer in the beads.
The extrusion-dripping method is commonly used to produce alginate beads because it is simple and inexpensive. Briefly, an alginate solution is extruded through a nozzle and allowed to break away from the nozzle to form droplets under the influence of gravitational force. Upon dripping, the droplets are nearly round (i.e., teardrop shaped), and they rapidly transform into round droplets due to the surface tension effect while falling, prior to hitting the gelling bath.18 The fabrication of alginate beads using extrusion-dripping method have been extensively studied by Chan et al. (2009).18 The production of uniform spherical beads is desirable, especially for drug delivery applications, because beads with a well-defined geometry allow precise and controllable drug release profile. However, until now, the fabrication of alginate beads with extremely high concentration remains a challenge due to the difficulties in producing spherical beads using the existing extrusion-dripping method.
In this study, we attempted a facile approach to develop calcium alginate beads with extended dissolution profile by increasing the alginate concentration above the viscosity limit (i.e., 5000 mPa s) that can be handled using the existing extrusion-dripping system. We developed a temperature-controlled extrusion-dripping system and studied its feasibility in forming spherical calcium alginate beads at ultra-high concentration (UHC) ranging from 6% w/v to 10% w/v, which corresponded to viscosity ranging from 33000 mPa s to 353000 mPa s. The operating temperature was varied between 40 °C and 80 °C. The effects of temperature and the heating duration on the solution viscosity and alginate depolymerization were first investigated. Next, the internal structure of the UHC calcium alginate beads was studied and compared with that of beads made using the normal alginate concentration (i.e., 2% w/v). Finally, the dissolution rate of the UHC calcium alginate beads in phosphate buffer solution was determined.
2 Materials and methods
2.1 Materials
Manugel GHB sodium alginate with an M/G ratio of 0.59 was used in this study (FMC Biopolymer, U.K., batch no. G5707201). A medium molecular-weight alginate was selected because this material is commonly used for encapsulation applications. Calcium chloride dihydrate, sodium chloride and hydrochloric acid were purchased from Fisher Scientific, UK. Monobasic sodium phosphate and dibasic sodium phosphate were purchased from Sigma. Vegetable oil was used as immiscible phase for the interphase column.2.2 Preparation of alginate solution
Alginate solutions with concentrations ranging from 2% w/v to 10% w/v were prepared by dispersing sodium alginate in distilled water. An alginate solution with a concentration of 2% w/v was used as the reference solution because this concentration is typically used for encapsulation studies. The solutions were mixed for 1 h at 1000 rpm using a mechanical stirrer.2.3 Determination of alginate solution properties
The freshly prepared alginate solutions were loaded into 100 ml Schott bottles. The bottles were immersed in a water bath and heated to between 40 °C and 80 °C for a period of up to 5 h. The Schott bottles were tightly sealed during the heating process to prevent water evaporation.ηr/c, which was based on the Kraemer equation, as follows:|
lnηr/c = [η] + k′[η]2c
| (1) |
| [η] = KMva | (2) |
2.4 Bead-fabrication device
Fig. 1 shows the custom-made device that was used to fabricate UHC alginate beads using the extrusion-dripping method. A water-jacketed vessel (1) was used to store the alginate solution, which was heated by circulating hot water in the water jacket. The solution temperature was controlled by regulating the temperature of the water circulating through a water bath. The temperature of the alginate solution in the vessel was continuously monitored using a temperature indicator. A pressure controller, which was connected to an air compressor (2), was used to control the rate of flow of the alginate solution through a 0.4 mm nozzle attached to the extrusion chamber (3). The piping was insulated to prevent heat loss. The extrusion chamber was fitted with two 240 V spotlights (4) that functioned as the heat sources. The temperature in the chamber was maintained at 60 °C or 80 °C by regulating the power supplied to the spotlights. The dimension of the extrusion-dropping chamber was 34 cm (width) × 40 cm (depth) × 85 cm (height), and it was insulated with an aluminum sheet to reduce heat loss. The droplets were dropped into a 0.6 M calcium chloride gelling bath at 25 °C (5) and stirred gently for 24 h. The distance between the nozzle tip and the gelling bath was set at 145 cm, and the effective falling distance of the heated solutions was approximately 70 cm.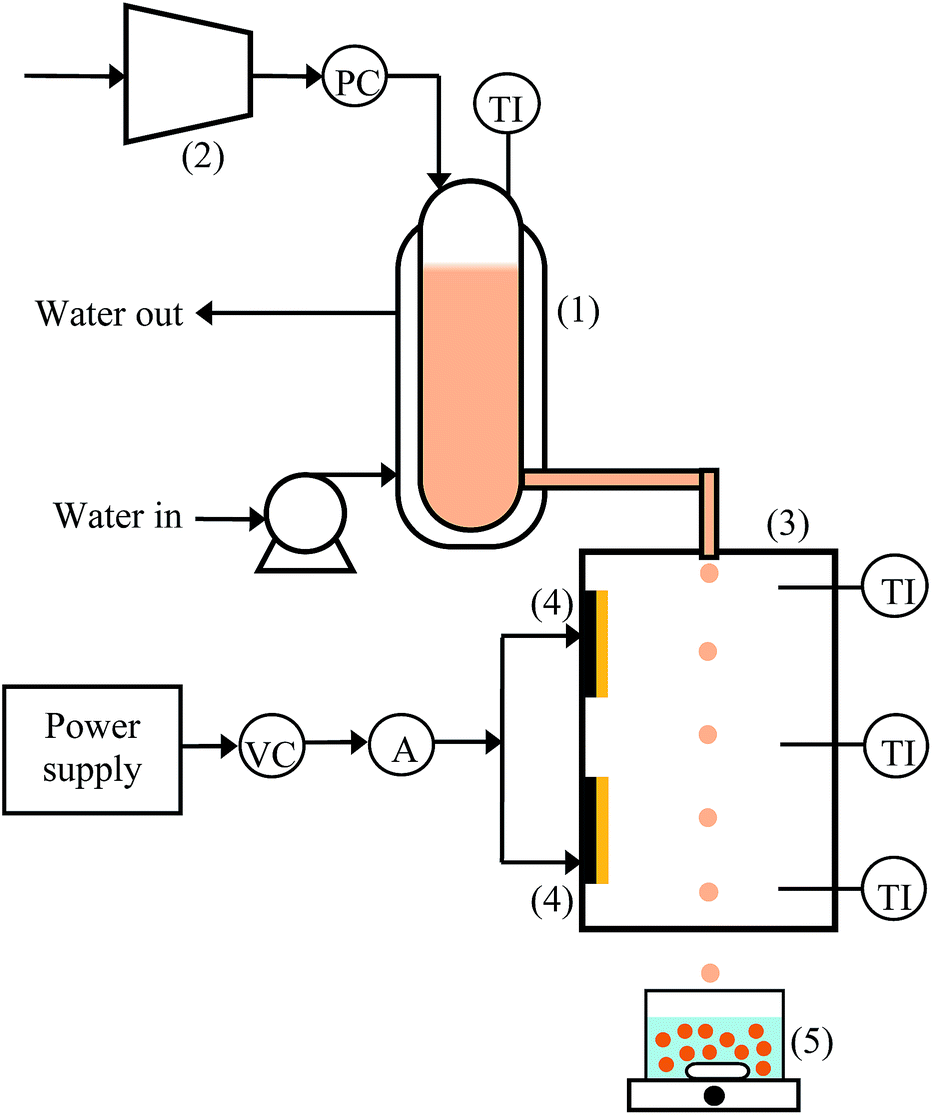 | ||
| Fig. 1 Experimental device for the fabrication of UHC alginate beads. (1) Water-jacketed vessel, (2) air compressor, (3) extrusion chamber, (4) heater, (5) gelling bath and magnetic stirrer. PC: pressure controller, TI: temperature indicator, VC: voltage controller, A: ammeter. | ||
2.5 Determination of minimum falling distance, Dmin
The relationship between the solution viscosity and Dmin required to form round beads using the extrusion-dripping method is shown in the following equation:| Dmin = 1.63e0.12Oh | (3) |
2.6 Characterization of calcium alginate beads
| SF = (dmax − dper)/(dmax + dper) | (4) |
3 Results and discussion
3.1 Viscosity of the alginate solutions
The viscosity of the unheated alginate solutions was measured at 25 °C. As expected, the viscosity increased exponentially as the alginate concentration increased. The viscosity of the alginate solutions at 2% w/v, 6% w/v, 8% w/v and 10% w/v was 630 mPa s, 33000 mPa s, 113000 mPa s and 353000 mPa s, respectively. The increase in the solution viscosity as the alginate concentration increased could be due to an increase in the extent of entanglement of the polymer chains and an increase in the extent of inter- and intramolecular interactions (i.e., hydrogen bonds) of the alginate molecules.21–23
3.2 Effect of heating on viscosity
Alginate solutions with concentrations of 2% w/v, 6% w/v, 8% w/v and 10% w/v were heated to a temperature between 40 °C and 80 °C and the solution viscosity was measured (see Fig. S1†). In general, the viscosity of alginate solutions between 2% w/v and 10% w/v reduced substantially when temperature increased. The viscosity of solutions reached a plateau after 1 h of heating and it decreased only slightly when the solutions were heated further.Interestingly, the viscosity of the alginate solutions within this concentration range was found to decrease by a same ratio (η/η0) when heated to a same temperature. Fig. 2a shows the normalized viscosity (η/η0) of the alginate solutions as a function of alginate concentration. The deviation of the viscosity ratios within the 5 h of heating was indicated by the error bars. Fig. 2b shows the average reduction in the viscosity ratio of the alginate solutions at different temperatures after heating for 5 h. The viscosity was found to decrease linearly by 43%, 58%, 71% and 90% irrespective of the alginate concentration when heated to 40 °C, 50 °C, 60 °C and 80 °C, respectively.
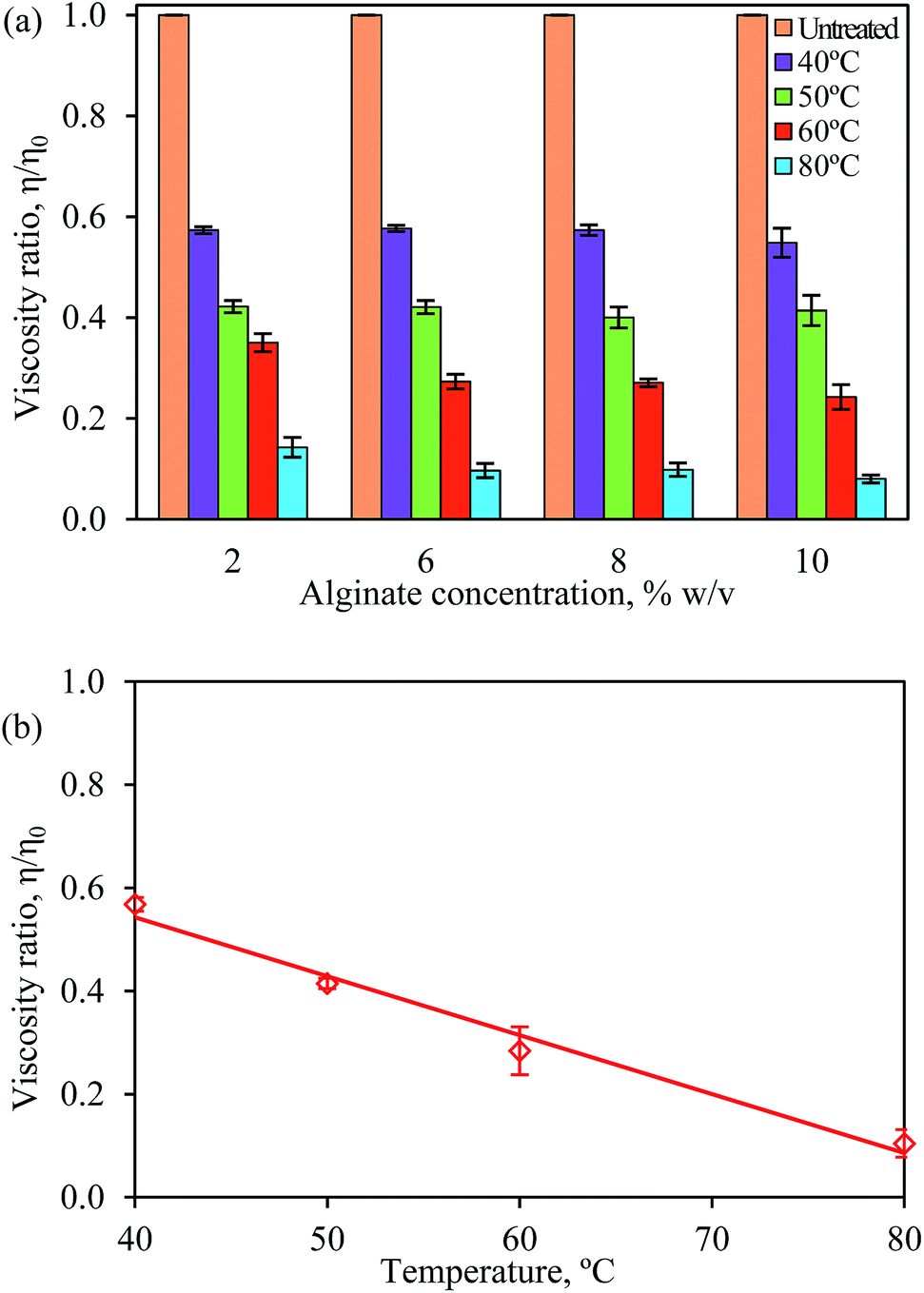 | ||
| Fig. 2 (a) Normalized viscosity (η/η0) of the alginate solutions as a function of alginate concentration after heating between 1 h to 5 h. The η0 for 2% w/v, 6% w/v, 8% w/v and 10% w/v alginate solutions are 630 mPa s, 33000 mPa s, 113000 mPa s and 353000 mPa s, respectively. (b) Average reduction in the viscosity ratio of alginate solutions (for all concentrations) at different temperatures after heating for 5 h. Error bars show the standard deviation of the mean of duplicate experiments. | ||
There are two possible explanations for the viscosity decreasing as the temperature increased. First, the thermal energy may have caused the rearrangement of the alginate molecules, resulting in the dissociation of the entangled chain and an increase in the inter- and intramolecular distances of the alginate molecules.22 Second, heating may have caused the cleavage of the glycosidic bonds that linked alginate monomers and the breakage of the polymer chains into shorter chains.24
3.3 Effect of heating on thermal depolymerization
The effect of heating on the degree of depolymerization of alginate macromolecules was determined based on the changes in the weight average molecular weight, Mw, of alginate with respect to that in the unheated sample, M0. The Mw was determined using the Mark–Houwink equation (eqn (2)) based on the intrinsic viscosity, which obtained from the Kraemer plot (see Fig. 3) by extrapolating the lines to an alginate concentration of zero. In the unheated alginate solution, which had an intrinsic viscosity of 528 ml g−1, alginate had a Mw of 264 kDa. When the solution was subjected to heating, the Mw of alginate decreased, which indicated the depolymerization of the alginate macromolecules.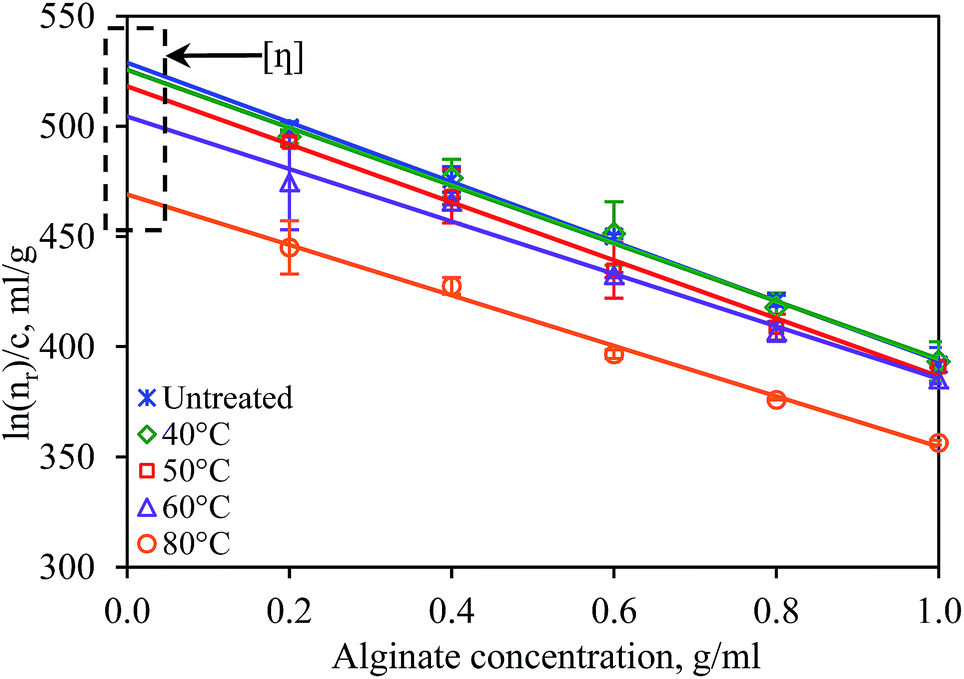 | ||
| Fig. 3 Changes in the intrinsic viscosity, [η], of 10% w/v alginate solutions after heating for 5 h between 40 °C and 80 °C. Error bars show the standard deviation of the mean of duplicate experiments. | ||
Fig. 4a shows the effects of temperature and the alginate concentration on the Mw of alginate. The changes in the Mw of alginate in 2% w/v and 10% w/v alginate solutions were compared after the solutions were heated for 5 h. The Mw of alginate in both 2% w/v and 10% alginate solutions was found to change with a similar trend at temperatures between 40 °C and 80 °C. No changes were found when the solutions were heated to 40 °C. Above this temperature, the Mw of alginate changed marginally, by 3% and 10% at 60 °C and 80 °C, respectively.
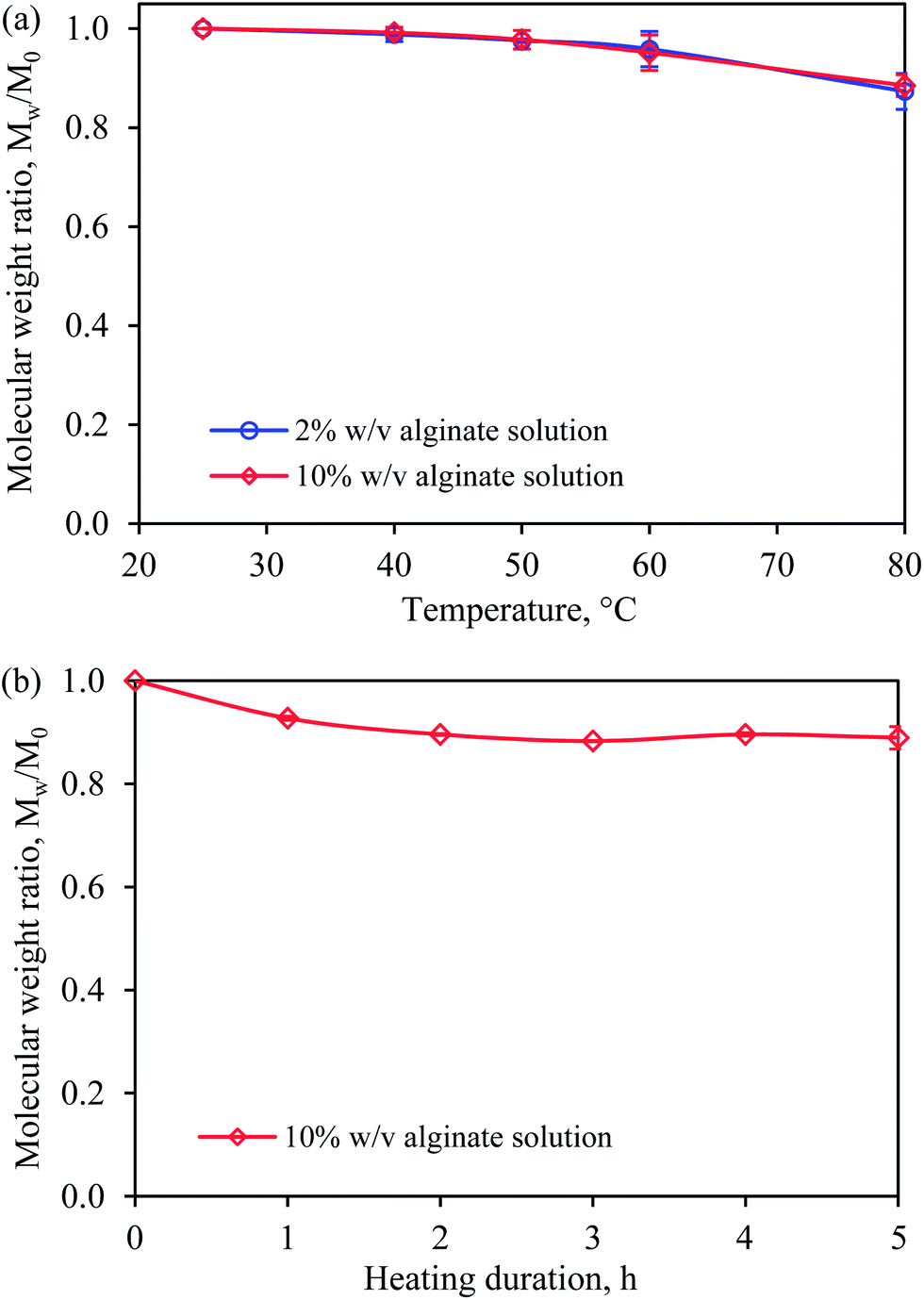 | ||
| Fig. 4 Changes in weight average alginate molecular weight, Mw, of (a) 2% w/v and 10% w/v alginate after heating for 5 h between 40 °C and 80 °C and (b) 10% w/v alginate after heating for five subsequent hours at 80 °C. Error bars show the standard deviation of the mean of duplicate experiments. | ||
The effect of the heating duration at 80 °C on the Mw of alginate in a 10% w/v alginate solution was investigated (see Fig. 4b). The Mw decreased slightly during the first 2 h of heating and no further reduction was observed after that point. The results obtained from the viscosity and Mw studies confirmed the feasibility of using a temperature-controlled extrusion method to fabricate UHC alginate beads because the solution viscosity could be substantially reduced at higher temperatures with only small changes in the Mw of alginate, which indicated little depolymerization of the alginate polymer.
3.4 Fabrication of UHC calcium alginate beads
The fabrication of calcium alginate beads using highly viscous solutions with a viscosity of up to 353000 mPa s was performed at an operating temperature of 60 °C or 80 °C. The temperatures in the water-jacketed vessel and the extrusion chamber were identical throughout the entire process to avoid heat loss from the droplets when they were extruded. The droplets were dropped into the gelling bath.
Fig. 5 shows images of a 10% w/v alginate solution that was extruded through the nozzle at an operating temperature of 80 °C. The extruded alginate liquid formed a long neck, resulting in a long breakup distance of 7.6 cm (see Fig. 5a). After breakup, the liquid droplet had a tail that was approximately 2 cm long (see Fig. 5b). When the operating temperature was lowered to 60 °C, the breakup distance and length of the tail on the droplets were longer due to the higher viscosity of the solution (images not shown). However, when a lower alginate concentration (i.e., 6% w/v) was used, the breakup distance and the tail were shorter due to the lower viscosity of the solution (i.e., breakup distance = 2 cm and tail length = 0.7 cm at an operating temperature of 80 °C). These droplets passed through a temperature-controlled extrusion chamber before contacting the gelling bath.
 | ||
| Fig. 5 Extrusion of the 10% w/v alginate solution through a nozzle at 80 °C (a) before droplet breakup and (b) after droplet breakup heat loss from the droplets when they were extruded. The droplets were dropped into the gelling bath. | ||
3.5 Bead shape
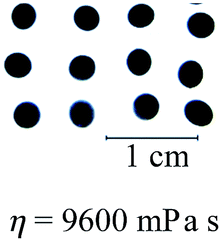
The alginate concentration and the operating temperature affected the shape of the beads formed, as shown in Table 1. Spherical calcium alginate beads, which had an SF of 0.04, were successfully fabricated using the 6% w/v alginate solution, which had a viscosity of 9600 mPa s, at an operating temperature of 60 °C. As expected, increasing the temperature to 80 °C improved the roundness of the beads (SF = 0.03) due to the lower viscosity of the solution at the higher temperature (i.e., 3300 mPa s).| Alginate concentration | Operating temperature | |
|---|---|---|
| 60 °C | 80 °C | |
| 6% w/v (η0 at 25 °C = 33000 mPa s) |
 |
 |
| 8% w/v (η0 at 25 °C = 113000 mPa s) |
 |
 |
| 10% w/v (η0 at 25 °C = 353000 mPa s) |
 |
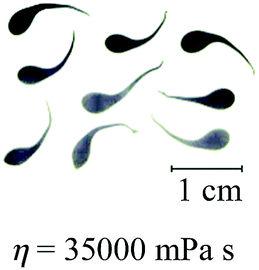 |
When the viscosity of the solution exceeded 9600 mPa s, calcium alginate beads with a distinct tail were formed (see Table 1). As expected, calcium alginate beads with a longer tail were formed when the solution became more viscous at a higher alginate concentration (i.e., 10% w/v versus 8% w/v) or a lower operating temperature (i.e., 60 °C versus 80 °C). Alginate solutions of higher viscosity or concentration formed droplets with a longer tail due to the increase of resistance to transforming into round droplets as they fall. The detached liquid droplets did not have sufficient time to transform into round droplets before contacting the gelling bath. In other words, the falling distance was insufficiently long for the completion of the shape-transformation process.
In previous study, the solution with the highest viscosity that was suitable for the fabrication of round calcium alginate beads using the extrusion-dripping method had a viscosity of approximately 5000 mPa s, which set the upper limit in the Oh–Re plot at Oh = 11 at 25 °C, as shown in Fig. 6.18 Solutions of a higher viscosity were not tested in their study because of the difficulty in pumping a viscous liquid using a peristaltic pump. In this investigation, a pressurized-air system was used to move the viscous solutions. This system allowed the precise control of the liquid flow rate through regulating the air pressure. Furthermore, the liquid could be extruded without pulsation, which is ideal for the production of mono-dispersed droplets. In this study, we found that round beads could be produced using a liquid with a viscosity as high as 9600 mPa s at 60 °C, which gave an Oh value of 27 (see Fig. 6).
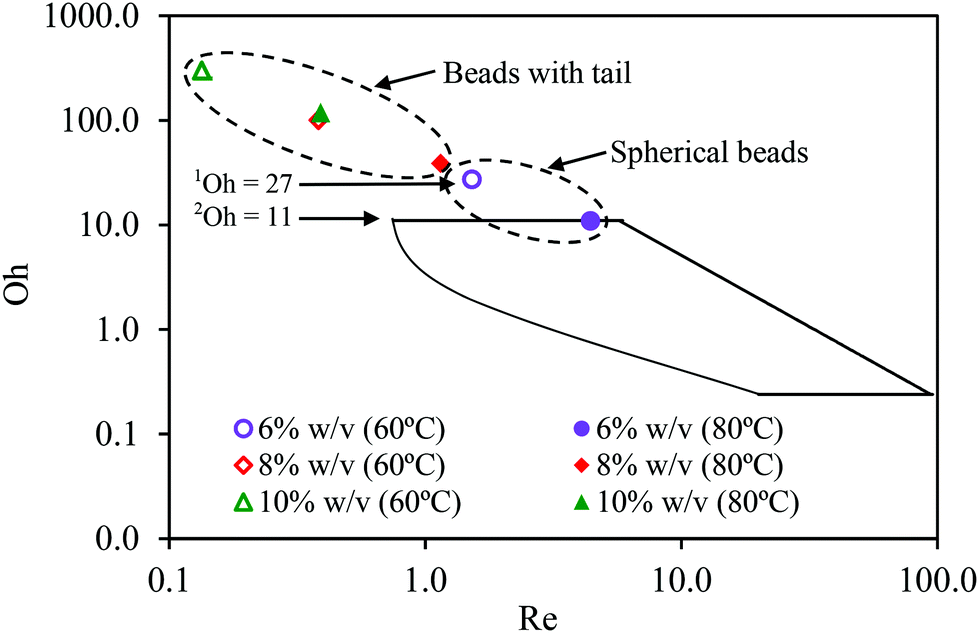 | ||
| Fig. 6 Oh–Re plot adopted from Chan et al. (2009). The boundaries surrounded by solid lines indicate the operating limits for the production of spherical beads.10 1Viscous limit 9600 mPa s at 60 °C obtained in this work, Oh = 27. 2Viscous limit 5000 mPa s at 25 °C obtained from Chan et al. (2009), Oh = 11. | ||
Eqn (3) states that the Dmin increases exponentially with Oh. When the alginate concentration is increased, the solution viscosity increases exponentially. However, the surface tension of the solution varies only marginally in relative to the solution viscosity.18 Therefore, Dmin is predominantly affected by the solution viscosity. The correlation between the Dmin and solution viscosity appropriate for the production of spherical beads can be expressed using eqn (3).
In this study, the falling distance was set at 70 cm. In theory, this falling distance could be used for the production of spherical beads using solutions with a viscosity of up to 9900 mPa s. The experimental results obtained in this study are in good agreement with the theoretical estimation because spherical beads produced using a 6% w/v alginate solution were found to have a viscosity of 3300 mPa s at 80 °C or 9600 mPa s at 60 °C (see Table 1). Using eqn (3), we subsequently estimated the falling distance as a function of the solution viscosity (see Fig. 7). In reality, the falling distance is typically limited by the ceiling height of a building. Assuming that the ceiling height of a typical factory is approximately 7 m, the maximal falling distance that can be set up for the extrusion process is approximately 5 m. At this height, the theoretical maximum viscosity of a solution that could be processed using this method would be approximately 15100 mPa s. This viscosity corresponds to a maximal alginate concentration of 8% with an initial viscosity of approximately 113000 mPa s at 25 °C. When this solution is heated to 80 °C, the viscosity will decrease by 90% and thus, in theory, this solution allows the formation of round beads.
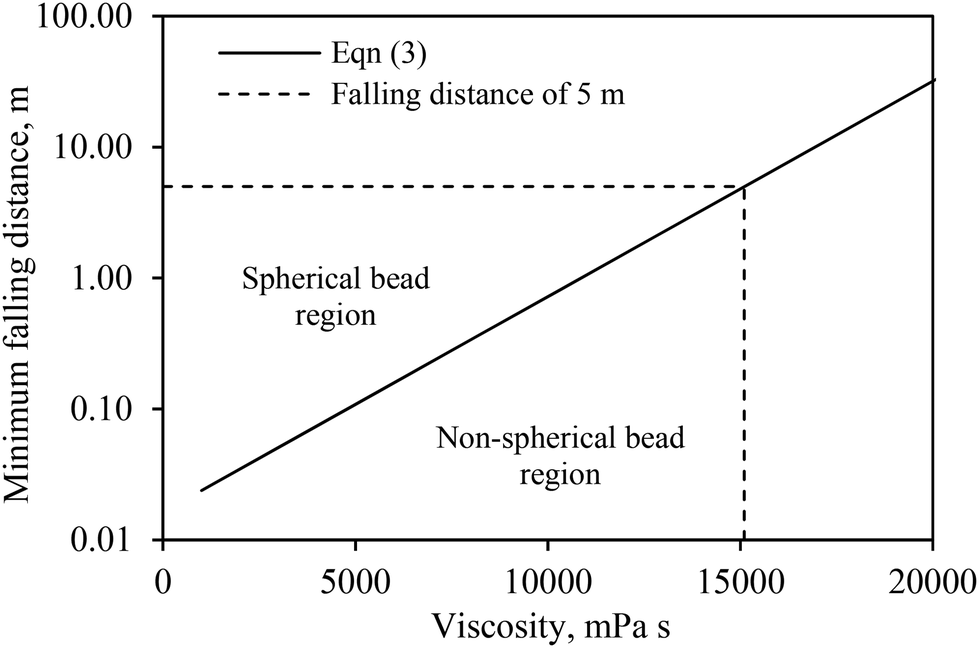 | ||
| Fig. 7 Correlation between minimum falling distance, Dmin, and solution viscosity for the production of spherical calcium alginate beads. | ||
3.6 FTIR spectroscopic analysis
The effect of heating on the properties of alginate was investigated by identifying the changes in the chemical bonds in alginate molecules using FTIR spectroscopic analysis. Fig. 8 shows the FTIR spectra of sodium alginate powder and calcium alginate beads (produced using 2% w/v or 6% w/v alginate) that were fabricated at 80 °C. The peaks present between 3400 cm−1 and 3200 cm−1 in the spectra represent the stretching vibration of the O–H bonds of alginate. The two band peaks at 1594 cm−1 and 1409 cm−1 were identified as representing the asymmetric and symmetric stretching vibrations, respectively, of the C–O bond of the COO− group. The shift from 1594 cm−1 to 1593 cm−1 and from 1409 cm−1 to 1417 cm−1 in the spectra of sodium alginate powder vs. calcium alginate beads was due to the replacement of the Na+ in the uronic-acid residues by Ca2+ and, hence, a change in the charge density and the radius of the atomic weight of the cation.25 The shift in the peak representing the C–O bond in the two spectra indicated the involvement of the COO− group in the Ca2+-mediated processes of alginate reticulation and egg-box-structure formation.26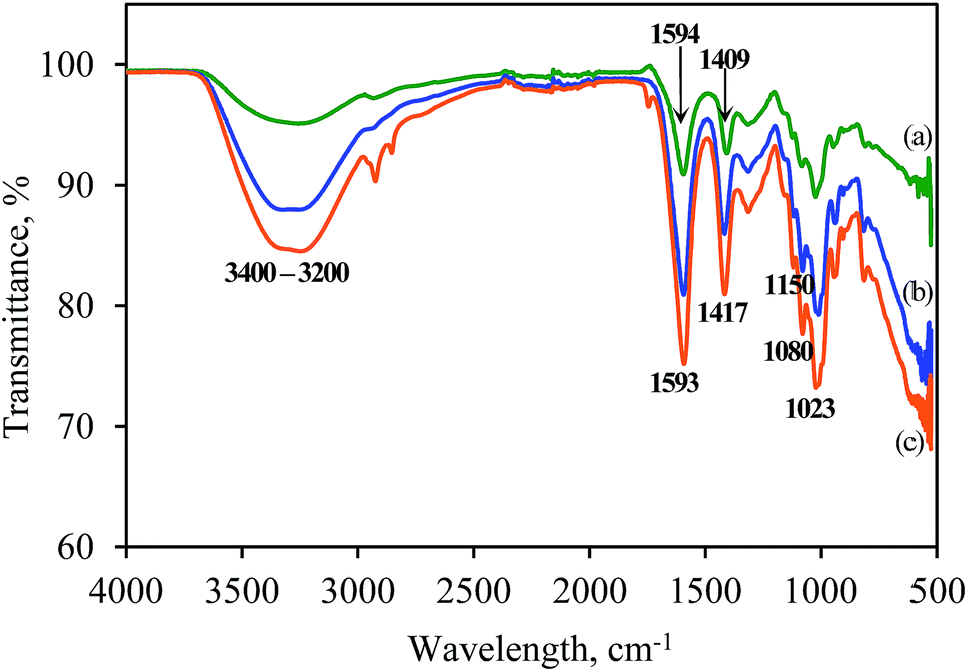 | ||
| Fig. 8 FTIR spectra of (a) sodium alginate powder, (b) 2% w/v calcium alginate beads and (c) 6% w/v calcium alginate beads fabricated at 80 °C. | ||
In contrast, no difference in the band peak at 1150 cm−1 that represents the asymmetric C–O–C stretching vibration was observed in the two spectra, which indicated that the glycosidic linkages among the alginate molecules was unchanged by gelation. In addition, the band peaks at 1080 cm−1 and 1023 cm−1, which relate to the stretching vibrations of the C–O bond of the glycosidic linkage, was unchanged by gelation at high operating temperature. Generally, the spectrum of the 10% w/v beads exhibited a lower percentage of transmittance compared with that of the 2% w/v beads. This result is due to the presence of more polymers in the 10% w/v beads, which could have restricted the transmittance of light.
3.7 FESEM analysis
Fig. 9 shows the FESEM images of cross-sections of dried calcium alginate beads. To preserve the original structure of the beads, they were freeze-dried prior to sectioning. The internal structures of the 2% w/v and 6% w/v beads were different in terms of their cavity sizes and cavity distributions. Fig. 9a shows that the internal structure of a 2% w/v bead resembled an interconnected mesh structure with many hollow cavities (dark areas). The interconnected network could have formed due to interchain interactions between the MG-blocks, which occur via carboxylate–Ca2+–carboxylate bridging, resulting in an abrupt change of the angle of orientation of the polymer molecules.27 | ||
| Fig. 9 FESEM images of calcium alginate beads cross-section: (a) 2% w/v bead, (b), (c) and (d) 6% w/v bead. | ||
The hollow cavities resulted from the elimination of the water in the hydrogels, which was removed under vacuum during the freeze-drying process. The shape and size of the cavities corresponded to the pouches of water that had been present in the beads. Small randomly and densely distributed cavities were observed within the calcium alginate network near the surface of the beads. However, the cavities were larger toward the core, which displayed a collapsed network structure, indicating the inhomogeneity of the gel structure. The inhomogeneous gel structure in calcium alginate beads is well known, and this process is caused by the gelling from the surface towards the inner core.28
In contrast, the 6% w/v beads had a multi-layered internal structure with large hollow cavities dispersed among distinct thick layers of calcium alginate matrix (see Fig. 9b). These cavities were more uniformly distributed within the 6% w/v beads than were those of 2% w/v beads, indicating that the former had a more homogeneous gel structure. This more homogeneous structure could be due to the high viscosity of the alginate solution used, which hindered the movement of alginate molecules and their diffusion to the surface of the droplets during gelation. Under this condition, gelation depended predominantly on the diffusion of Ca2+ to the gelling zone of the alginate polymers.
It is worth noting that the first layer of the calcium alginate matrix formed at the surface of the 6% w/v bead was very thick (i.e., 40 μm) compared to that of 2% w/v bead (i.e., 2 μm). The thick matrix layer on the surface of 6% w/v bead resembled a distinct shell enveloping the other calcium alginate matrix layers. The enlarged views of the shell layer, shown in Fig. 9c and d, reveal that the thick shell layer is composed of multiple layers of calcium alginate matrix that are interconnected by fine strands. The bulky layers of calcium alginate matrix at the surface of the beads formed easily at the beginning of the gelation process due to abundance of Ca2+ and alginate available for crosslinking. Whereas the cross-linking process in the interior of the beads occurred very slowly due to the diffusion resistance of Ca2+ into this region. The slow gelation process due to the limited amount of free Ca2+ in the interior of the beads could result in the alginate molecules having more time to rearrange and bind to each other, thus forming a thick matrix layer. After the cross-linking sites in a particular layer were depleted, a new layer would be formed. This phenomenon could explain the existence of large cavities between the calcium alginate matrix layers.
3.8 Dissolution of calcium alginate beads
The dissolution of calcium alginate beads at 2% w/v and 6% w/v was performed in HCl solution (pH 1.2) and phosphate buffer solution (pH 7.4), mimicking the gastric and intestinal fluids, respectively. The beads were found to remain intact after incubation in the HCl solution for 2 h (see Fig. S2 and S3†). However, the beads were dissolved in the phosphate buffer solution. The dissolution rate of the beads was determined based on the release rate of the divalent cations, Ca2+ ions, from the beads.Fig. 10 shows the dissolution rate of the calcium alginate beads in the 0.1 M phosphate-buffer solution (pH 7.4). For the 2% w/v calcium alginate beads, the release of Ca2+ ions occurred rapidly and completed after 80 min. The release of all the bound Ca2+ ions to the bulk medium indicated the complete dissolution of the calcium alginate hydrogel matrices. Similarly, the release of Ca2+ ions from the 6% w/v calcium alginate beads occurred rapidly within 20 min, followed by a gradual release and completed after 240 min. The beads were found to swell while the Ca2+ ions were released gradually and then slowly dissolved.
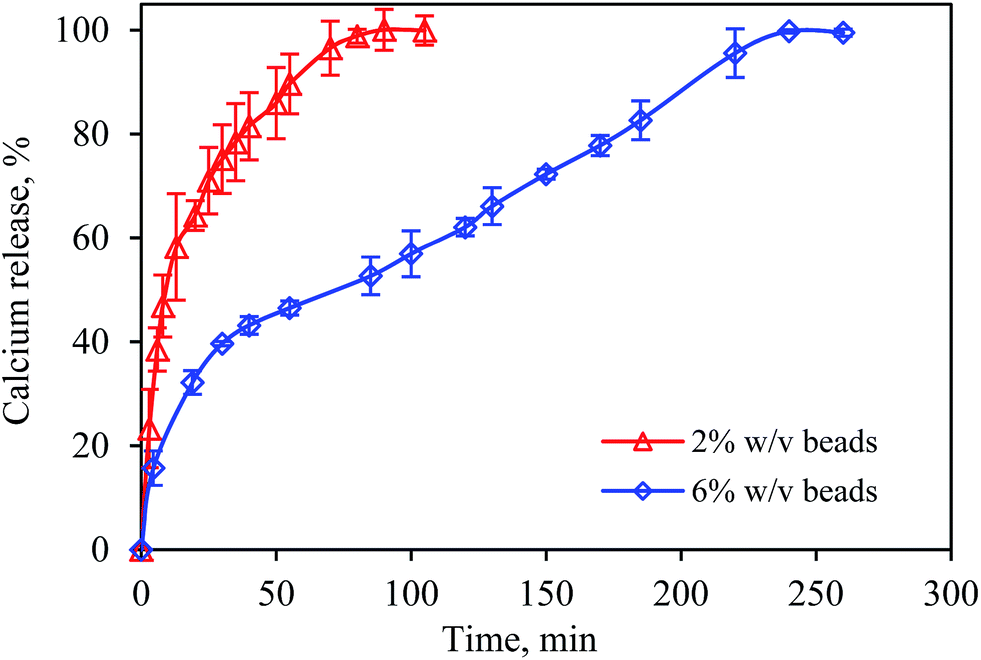 | ||
| Fig. 10 Release profile of calcium ions from calcium alginate beads in phosphate buffer solution at pH 7.4 and 37 °C. Error bars show the standard deviation of the mean of duplicate studies. | ||
The dissolution of beads was attributed to ion exchange between the Na+ ions in the phosphate-buffer solution and the Ca2+ ions that were bound to alginate.29 During the ion exchange process, the content of free carboxylate anions in the gel increased, leading to electrostatic repulsion between alginate chains, which ultimately caused chain relaxation and promoted gel swelling.9,30 The beads began to disintegrate when the calcium alginate networks were unable to retain the structure and finally, the beads dissolved.
This study demonstrated that the dissolution time increased with alginate concentration. The 2% w/v beads were completely dissolved after 80 min, whereas the dissolution time for 6% w/v beads was three times longer (i.e., 240 min). For beads with a higher alginate concentration, the ion-exchange process was longer because more Ca2+ ions were bound to the alginate. In addition, diffusion resistance could have occurred and slowed down the diffusion of Na+ ions into the high-density gel to exchange with Ca2+ ions.
4 Conclusions
This study shows that it is feasible to produce UHC calcium alginate beads with extended dissolution profile using a temperature-controlled extrusion method. The viscosity of the alginate solutions was significantly reduced between 40 °C and 80 °C, and the viscosity-reduction ratio was linearly proportionally to the temperature but was independent of the alginate concentration. Heating the alginate solutions was found to have a marginal effect on alginate depolymerization. Spherical UHC calcium alginate beads were successfully produced using an alginate solution with an initial viscosity of 33000 mPa s. The FTIR spectroscopic analysis revealed the process of producing the UHC calcium alginate beads did not change the chemical properties of alginate. Interestingly, the UHC calcium alginate beads had a multi-layered internal structure with distinct thick layers of calcium alginate matrix and large cavities dispersed among them. The dissolution time for the UHC calcium alginate beads in the phosphate buffer solution was three times longer than that of the normal beads (i.e., 2% w/v). The ease of production of the UHC beads and their unique properties indicate their strong potential for applications in sustained-release of food and pharmaceutical products, which release from the systems via chemical or physical breakdown of the matrix.
Acknowledgements
The authors wish to thank the Ministry of Higher Education of Malaysia for supporting this investigation under the FRGS/1/2013/TK04/MUSM/02/1 scheme and the School of Engineering of Monash University, Malaysia for providing a PhD scholarship to Wan-Ping Voo.Notes and references
- A. Haug, B. Larsen and O. Smidsrød, Acta Chem. Scand., 1966, 35, 183–190 CrossRef PubMed.
- G. T. Grant, E. R. Morris, D. A. Rees, P. J. C. Smitch and D. Thom, FEBS Lett., 1973, 32, 195–198 CrossRef CAS.
- W. P. Voo, P. Ravindra, B. T. Tey and E. S. Chan, J. Biosci. Bioeng., 2011, 111, 294–299 CrossRef CAS PubMed.
- S. Y. Ahn, C. H. Mun and S. H. Lee, RSC Adv., 2015, 5, 15172–15181 RSC.
- J. Zhao, B. Guo and P. X. Ma, RSC Adv., 2015, 4, 17736–17742 RSC.
- S. Martins, B. Sarmento, E. B. Souto and D. C. Ferreira, Carbohydr. Polym., 2007, 69, 725–731 CrossRef CAS PubMed.
- A. Kikuchi, M. Kawabuchi, M. Sugihara, Y. Sakurai and T. Okano, J. Controlled Release, 1997, 47, 21–29 CrossRef CAS.
- Y. Murata, K. Nakada, E. Miyamoto, S. Kawashima and S. H. Seo, J. Controlled Release, 1993, 23, 21–26 CrossRef CAS.
- A. Kikuchi, M. Kawabuchi, A. Watanabe, M. Sugihara, Y. Sakurai and T. Okano, J. Controlled Release, 1999, 58, 21–28 CrossRef CAS.
- J. L. West, Nat. Mater., 2003, 2, 709–710 CrossRef CAS PubMed.
- S. Y. Lin and J. W. Ayres, Pharm. Res., 1992, 9, 1128–1131 CrossRef CAS.
- M. L. Huguet, N. J. Neufeld and E. Dellacherie, Process Biochem., 1996, 31, 347–353 CrossRef CAS.
- T. Agarwal, S. N. G. H. Narayana, K. Pal, K. Pramanik, S. Giri and I. Banerjee, Int. J. Biol. Macromol., 2015, 75, 409–417 CrossRef CAS PubMed.
- P. Eiselt, K. Y. Lee and D. J. Mooney, Macromolecules, 1999, 32, 556–5566 CrossRef.
- J. Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- M. Leonard, M. Rastello De Boisseson, P. Hubert, F. Dalençon and E. Dellacherie, J. Controlled Release, 2004, 98, 395–405 CrossRef CAS PubMed.
- B. L. Yao, C. H. Ni, C. Xiong, C. P. Zhu and B. Huang, Bioprocess Biosyst. Eng., 2010, 33, 457–463 CrossRef CAS PubMed.
- E. S. Chan, B. B. Lee, P. Ravindra and D. Poncelet, J. Colloid Interface Sci., 2009, 338, 63–72 CrossRef CAS PubMed.
- E. O. Kraemer, Ind. Eng. Chem. Res., 1938, 30, 1200–1203 CrossRef CAS.
- O. Smidsrød and A. Haug, Acta Chem. Scand., 1968, 22, 797–810 CrossRef PubMed.
- E. R. Morris, A. N. Cutler, S. B. Ross-Murphy and D. A. Rees, Carbohydr. Polym., 1981, 1, 5–21 CrossRef CAS.
- A. Watthanaphanit and N. Saito, Polym. Degrad. Stab., 2013, 98, 1072–1080 CrossRef CAS PubMed.
- R. Kohn, Pure Appl. Chem., 1975, 42, 371–397 CrossRef CAS.
- T. M. Aida, T. Yamagata, M. Watanabe and R. L. Smith Jr, Carbohydr. Polym., 2010, 80, 296–302 CrossRef CAS PubMed.
- H. Daemi and M. Barikani, Sci. Iran., 2012, 19, 2023–2028 CrossRef CAS PubMed.
- A. Dalmoro, A. Barba, G. Lamberti, M. Grassi and M. d'Amore, Adv. Polym. Technol., 2012, 31, 219–230 CrossRef CAS PubMed.
- H. L. Ma, X. R. Qi, Y. Maitani and T. Nagai, Int. J. Pharm., 2007, 222, 177–186 CrossRef PubMed.
- G. Skjåk-Bræk, H. Grasdalen and O. Smidsrød, Carbohydr. Polym., 1989, 10, 31–54 CrossRef.
- R. S. Giridhar and A. S. Pandit, Int. J. Polym. Mater., 2013, 62, 743–748 CrossRef CAS.
- S. K. Bajpai and S. Sharma, React. Funct. Polym., 2004, 59, 129–140 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03862f |
| This journal is © The Royal Society of Chemistry 2015 |