DOI:
10.1039/C5RA03771A
(Paper)
RSC Adv., 2015,
5, 30445-30455
Donor–acceptor copolymers containing the phthalazinone–thiophene structure synthesized by classical nucleophilic aromatic polymerization†
Received
3rd March 2015
, Accepted 13th March 2015
First published on 13th March 2015
Abstract
A series of new donor–acceptor copolymers based on a novel monomer containing phthalazinone and thiophene structures, were synthesized by classical nucleophilic displacement step polymerization reaction, and then were fully characterized. Their thermal, optical, electrochemical properties and quantum chemical calculations were investigated in detail. The comparisons between the linkage of ketone/di-ketone/sulfone groups and phenyl/naphthalene groups in the resulting polymer backbone were carried out to explore the relationship of the structure and the properties in the polymer system. The number average molecular weights of the resulting copolymers are 15 kDa–24 kDa. All of the copolymers are stable up to 441 °C, and their 5% weight loss temperatures are higher than 489 °C. Their optical band gap and HOMO energy levels vary from 2.36 eV to 2.76 eV and −5.16 eV to −5.51 eV, respectively. The soluble copolymers obtained exhibit blue-fluorescence. Their photoluminescence quantum yield ranges from 0.04 to 0.28 in N-methyl-2-pyrrolidone. These non-ether bond polymers may be potential candidates in photoelectric application.
Introduction
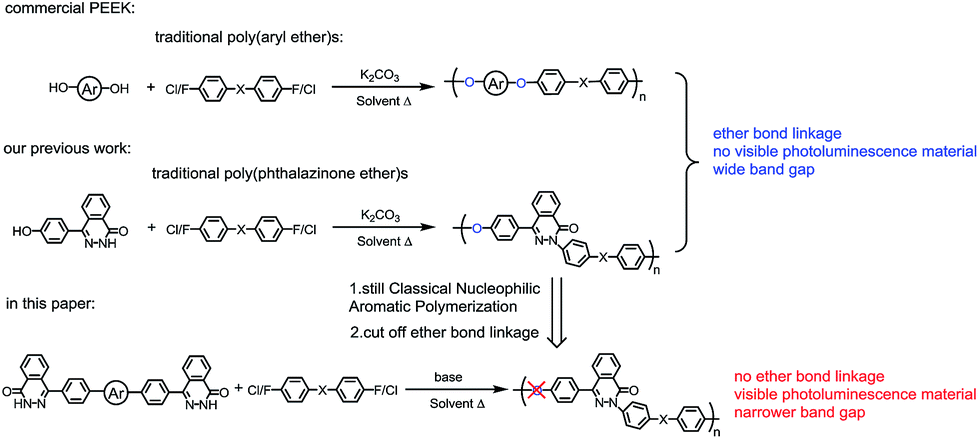
Conjugated polymers have attracted an increasing amount of attention in recent decades for various organic electronic devices such as organic light-emitting diodes (OLEDS), organic field-effect transistors (OFETs) and polymer solar cells (PSCs), because of their potential advantages over inorganic and small-molecule organic semiconductors.1 Polymeric materials usually have better film-forming ability, solution process ability, and mechanical properties.2 Researchers have been devoting themselves to develop cheap polymer materials having excellent photoelectric property.
Our group has been focusing on exploring heat resistant polymers based on the asymmetric monomer 1,2-dihydro-4-(4-hydrox-yphenyl)-1-(2H)-phthalazinone (DHPZ) (Fig. 1). As reported in our previous study, the NH group in DHPZ behaves like a phenolic OH group with the result that DHPZ can react with activated dihalo compounds to synthesize high molecular weight polymers via combined C–N and C–O condensation reactions.3 For example, poly(phthalazinone ether sulfone)s and poly(phthalazinone ether ketone)s were first successfully synthesized in 1993 by classical nucleophilic aromatic substitution reactions as shown in Scheme 1.4 The rigid asymmetric phenyl–phthalazinone structures in the polymer main chains improve the solubility of the resulting polymers with maintaining excellent thermostability, high glass transition temperature and mechanical properties.5–11 These high performance polymers have been widely used in industrial applications, such as optical wave guides films, ultrafiltration and nanofiltration membranes, and lithium-ion batteries functional films, anion exchange membrane materials used for vanadium redox flow battery applications, dielectric energy storage thin films, and proton exchange membrane materials for fuel cells.12–17 On the other side, DHPZ can be used as a acceptor due to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond and lactam of the phthalazinone structure.18 Also its sp3 hybrid nitrogen atom in DHPZ has better planarity than classical triphenylamine structure.19 The special structure of DHPZ makes it attractive for constructing n-type and donor–acceptor polymers. However, to our knowledge, few papers report about the applications of phthalazinone-containing polymers in optoelectronic materials. One of the main reasons is that the existence of ether bond in the polymer main chain interrupts conjugation effect. Thus, to eliminate the ether bond and prolong the conjugated bond length in the phthalazinone-containing polymers can broaden their application into optoelectronic materials field. The concept of our design is summarized in Scheme 1.
N bond and lactam of the phthalazinone structure.18 Also its sp3 hybrid nitrogen atom in DHPZ has better planarity than classical triphenylamine structure.19 The special structure of DHPZ makes it attractive for constructing n-type and donor–acceptor polymers. However, to our knowledge, few papers report about the applications of phthalazinone-containing polymers in optoelectronic materials. One of the main reasons is that the existence of ether bond in the polymer main chain interrupts conjugation effect. Thus, to eliminate the ether bond and prolong the conjugated bond length in the phthalazinone-containing polymers can broaden their application into optoelectronic materials field. The concept of our design is summarized in Scheme 1.
 |
| | Fig. 1 Energy-minimized conformation of DHPZ determined using DFT (calculated by Gaussian 09W at the B3LYP/6-31G**level). | |
 |
| | Scheme 1 Synthetic route of poly(phthalazinone ether)s. | |
In this paper, di-NH end-capped monomer M1 was designed and then synthesized (Scheme 2). Monomer M1 possesses several virtues: (1) its NH group can react with various dihalo-compounds (–F, –Cl, –Br, –I) catalyzed by low costly base through classical nucleophilic aromatic substitution polymerization. (2) By virtue of the CN bond and lactam structure, its polymers enrich the typical imide- and amide-functionalized polymer semiconductor family, which has the high electron deficiency and the capability to self-assemble into ordered structures.20 (3) Its derivative M2 from the reaction of M1 and 1-bromododecane greatly exhibits potential blue light emitting with 54% of photoluminescence quantum yield (PLQY), compared to bi(9,9-diarylfluorene)s with PLQY = 70% and bifluorene series compound with PLQY = 60%.21 (4)Its polymers also have excellent thermal and chemical stability. Thus herein the di-NH group of M1 polymerized with di-halogenated compounds by classical nucleophilic aromatic substitution polymerization to produce a series of “non-ether bond” polymers (Scheme 3). In order to comparison with traditional poly(phthalazinone ether)s and commercial poly(aryl ether)s synthesized by the same polymerization method,22 the common monomers used in poly(aryl ether)s like M3 ∼ M7 were chosen in our work. Also, weak conjugated groups such as ketone/di-ketone/sulfone groups give polymers, without alkyl substituents, enough solubility in organic solvent for spin-coating process. The thermal properties, optical properties, electrochemical properties, quantum chemical calculations (B3LYP/6-31G**) by Gaussian 09 package23 and physical properties of the resulting polymers were investigated in details. In addition, the comparisons between the linkage of ketone/di-ketone/sulfone groups and phenyl/naphthalene groups in the backbone of the resulting polymers were carried out to explore the relationship of structure and property in our polymer system.
 |
| | Scheme 2 Synthesis of Di-NH capped monomers M1 and M2. | |
 |
| | Scheme 3 Synthetic route and structures of copolymers. | |
Experimental
Materials
Unless otherwise indicated, starting materials were obtained from Aldrich or Alfa Aesar and were used without further purification. Solvent and other common reagents were obtained from Shanghai Energy Chemical. THF was distilled from benzophenone and sodium under an inert N2 atmosphere prior to use. Quinoline was purified by distillation under vacuum. Commercial cuprous chloride was dissolved in concentrated hydrogen chloride and the resulting solution was filtered through an acid funnel to remove the insoluble substance. The liquid obtained was then diluted by pure water then to collect the white precipitate. The white solid was washed by methanol and ether, respectively, and then kept in a desiccator. CuCl/quinolone catalyst was prepared as the literature reported.3
Methods instrumentation
1H-NMR (400 Hz) and 13C-NMR (100 Hz) spectra were obtained with a Brucker spectrometer at an operating temperature of 25 °C using CDCl3 as solvents. HPLC-MS analyses were performed on a HP1100LC/MSD instrument. Gel permeation chromatography (GPC) analysis was carried out on a HP 1090 HPLC instrument equipped with 5 μm phenogel columns (linear, 4 × 500 Å) arranged in series with chloroform as solvent and a UV detector at 254 nm. And the values were calibrated versus polystyrene standard. Matrix-assisted laser desorption ionization time-of-flight mass spectroscopy (MALDI-TOF-MS) analyses were performed on a Micromass GC-TOF CA 156 MALDI-TOF/MS. Infrared measurements were performed on a Thermo Nicolet Nexus 470 Fourier transform infrared (FT-IR) spectrometer. UV-visible absorption was measured with a PerkinElmer Lambda 35 UV-vis spectrometer. Fluorescence spectra were obtained using a Hitachi F-4500 spectrofluorometer with a xenon lamp and 1.0 cm quartz cells. Elemental analysis was measured on a Vario ELIII CHNOS Elementaranalysator from Elementaranalysesyteme GmbH. Thermogravimetric analysis (TGA) of the polymers were performed on a Mettler TGA/SDTA851 thermogravimetric analysis instrument in a nitrogen atmosphere at a heating rate of 20 °C min−1 from 25 to 800 °C. Decomposition temperature (Td) in nitrogen was taken as the temperature of 5% weight loss. Char yield (Cy) was calculated as the percentage of solid residue after heating from 25 to 800 °C in flowing nitrogen. The glass transition temperature (Tg) was determined with a Mettler DSC822 differential scanning calorimetry (DSC) in flowing nitrogen at a heating rate of 10 °C min−1 from 25 to 350 °C. The Tg value was taken at the inflection point. Cylic voltammetric experiments were carried out on an electrochemistry workstation with a BAS100W voltammetric analyzer. A three-electrode setup was performed in a dichloromethane containing 0.1 M tetrabutylammonium hexafluoroborate as the supporting electrolyte, used employing platinum working and counter electrodes and an Ag/Ag+ reference electrode (containing 0.01 M silver nitrate in dichloromethane). Potentials were recorded relative to the ferrocene/ferrocenium (Fc/Fc+) redox couple which occurs at a value of +0.07 V under these conditions. LUMO and HOMO energies were calculated from the onset of the first reduction peak assuming a formal potential of Fc/Fc+ of −4.80 eV relative to vacuum level (scan rate 50 mV s−1). The optical band gap was calculated from measured at the onset of absorption. Quantum yield measurements of the polymers, dissolved in organic solvent, were performed using quinine sulfate (quantum yield = 0.546, excited at 346 nm, in 0.1 N H2SO4) as standards.24 DFT calculations were performed using the Gaussian 09W program at the B3LYP level with a 6-31G(d,p) basis set.23
Synthesis of monomers and polymers
4,4′-(thiophene-2,5-diylbis(4,1-phenylene))bis(phthalazin-1(2H)-one) (M1). To a mixture of 2,5-bis(tributylstannyl)thiophene (66.22 mg, 0.1 mmol) and 4-(4-bromophenyl)phthalazin-1(2H)-one (60.22 mg, 0.2 mmol) in N-methyl-2-pyrrolidone (NMP) 10 mL was added PdCl2(PPh3)2 (2.8 mg, 0.4% mmol) under a N2 atmosphere. The mixture was stirred at 110 °C for 42 h. A green precipitate was filtered. The resulting solid was washed with hot N,N-dimethyl acetamide (DMAc), NMP and water, and subjected to Soxhlet extraction successively with ethanol and dried under vacuum. 31.0 mg of greyish-green product was obtained (yield: 75%). Anal. calcd for C32H20N4O2S: C, 73.27; H, 3.84; N, 10.68; S, 6.11. Found: C, 68.73; H. 3.81; N, 10.06; S, 5.88.
4,4′-(thiophene-2,5-diylbis(4,1-phenylene))bis(2-dodecylphthalazin-1(2H)-one) (M2). A 50 mL, 3-necked round-bottomed flask equipped with a Dean–Stark trap, condenser, inert gas inlet, magnetic stirrer, was charged with M1 (524.6 mg, 1 mmol), cesium carbonate (325.82 mg, 1 mmol), hexamethylphosphoric triamide (HMPA) (6 mL) and toluene (10 mL). The mixture was heated to 145 °C (oil bath) under an argon atmosphere and maintained at this temperature for 4 h to remove water produced during the reaction. After complete dehydration, the reaction temperature was increased to 190–195 °C (oil bath) and the toluene was distilled. The yellow slurry in the flask was cooled to 80 °C. And 1.6405 g (5 mmol) of 1-bromododecane (498.4 mg, 2 mmol) was carefully added. The reaction mixture was then heated to 190–195 °C. The Cu(I) Cl/quinoline catalyst (0.5 mL) was injected into the reaction mixture. The resulting mixture was maintained at this temperature for 17 h, after which time, the color of reaction solution became dark red, indicating that the potassium salt of M1 had almost gone into solution. Then the solution was poured into the water in the presence of some dilute HCl. The organic compounds were extracted with DCM. The organic layer was dried over Na2SO4 and concentrated in vacuo. The crude product was purified by column chromatography using n-hexane as the eluent. 697.6 mg of brown product was obtained (yield: 81%). 1H-NMR (400 MHz, CDCl3) δ 8.56 (dd, J = 7.8, 1.5 Hz, 1H), 7.87–7.72 (m, 5H), 7.66 (d, J = 8.3 Hz, 2H), 7.43 (d, J = 11.5 Hz, 1H), 4.36–4.25 (m, 2H), 1.91 (dt, J = 14.9, 7.6 Hz, 2H), 1.42–1.21 (m, 18H), 0.87 (t, J = 6.8 Hz, 3H). 13C-NMR (101 MHz, CDCl3) δ 158.98 (s), 146.24–146.04 (m), 143.42 (s), 134.90 (s), 134.53 (s), 132.73 (s), 131.36 (s), 130.14 (s), 128.97 (s), 128.35 (s), 127.31 (s), 126.50–126.16 (m), 125.81 (s), 124.84 (s), 51.51 (s), 31.94 (s), 29.56 (dd, J = 16.3, 11.9 Hz), 28.66 (s), 26.83 (s), 22.71 (s), 14.15 (s). MALDI-TOF [M + H]+: calc. As: C56H68N4O2S; 861.25 (m/z). Found: 861.46 (m/z).
General synthesis of poly(phthalazinone ether)s by classical nucleophilic aromatic substitution reactions
A typical synthetic procedure of PDSK82 was described as follows (shown in Scheme 3). A mixture of M1 (1.0852 g, 2 mmol), M3 (0.4594 g, 1.6 mmol), M4 (0.0087 g, 0.4 mmol), anhydrous Cs2CO3 (0.9775 g, 3 mmol), 20 mL HMPA and 40 mL toluene was placed in a 100 mL three-necked round-bottomed flask equipped with a mechanical stirrer, a nitrogen inlet and outlet. The reaction mixture was constantly stirred and heated to 142 °C for 3 h, maintained at this temperature for 4 h to remove water produced during the reaction. After complete dehydration, the reaction temperature was increased to 190–195 °C (oil bath) and the toluene was distilled. The resulting mixture was maintained at this temperature for 17 h. Then the reaction mixture was slowly poured into sufficient ethanol at the presence of hydrochloric acid in drops. The crude polymer was washed for six times with hot distilled water to remove inorganic salts. The dried polymer was purified by dissolving in NMP, being filtered through a 0.45 mm Teflon micro filter before pouring into ethanol, and subsequently washed six times with hot deionized water. The polymers were purified by Soxhlet extraction with ethanol and acetone sequentially. The purified polymer was dried at 120 °C under vacuum for 24 h. The product was obtained in yield 74% (1.11 g). 1H-NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.12–7.92 (m, 4H), 7.92–7.74 (m, 5H), 7.67 (s, 2H), 7.43 (t, J = 13.5 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ 175.04 (s), 158.72 (s), 147.79 (s), 145.81 (s), 143.22 (s), 139.54 (s), 135.14 (s), 133.67 (s), 132.03 (s), 130.33 (d, J = 40.2 Hz), 130.08 (s), 129.59 (s), 129.09 (s), 128.67 (s), 128.06 (d, J = 28.3 Hz), 127.92–127.65 (m), 126.84 (s), 125.60 (t, J = 34.6 Hz), 124.98 (s). GPC: Mn = 16.5 kDa, Mw = 54.5 kDa, PDI = 3.3.
Others copolymers with different monomers were prepared using the similar procedures as outlined above and characterized as follow.
PDS. Yellow product. Yield: 73%. 1H-NMR (400 MHz, CDCl3) δ 8.59 (s, 1H), 8.11–7.90 (m, 4H), 7.88–7.72 (m, 4H), 7.67 (s, 2H), 7.52 (d, J = 1.7 Hz, 1H), 7.41 (d, J = 7.5 Hz, 1H). 13C-NMR (101 MHz, CDCl3) δ 175.09 (s), 158.74 (s), 147.82 (s), 145.86 (s), 143.20 (s), 139.51 (s), 135.11 (s), 133.67 (s), 132.10–131.90 (m), 130.13 (s), 129.39 (s), 128.64 (s), 128.21 (s), 127.24–126.62 (m), 125.92 (s), 125.07 (s). GPC: Mn = 24.4 kDa, Mw = 92.7 kDa. PDI = 3.8.
PDSK55. Black product. Yield: 79%. Calc. for C455H300N40O30S15: C, 74.98; H, 4.15; N, 7.69; O, 6.59; S, 6.60. Found: C, 67.61; H, 3.60; N, 6.54; S, 5.68.
PDSKK82. Brown product. Yield: 79%. 1H-NMR (400 MHz, CDCl3) δ 8.57 (s, 1H), 8.13–7.90 (m, 4H), 7.90–7.46 (m, 9H), 7.41 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 175.19–175.05 (m), 158.80 (s), 145.90 (s), 143.31 (s), 139.78–139.51 (m), 135.46–135.05 (m), 133.74 (s), 132.11 (s), 129.91 (d, J = 48.9 Hz), 129.63–129.43 (m), 129.17 (s), 128.51 (d, J = 48.1 Hz), 127.99–127.84 (m), 127.55 (s), 126.93 (s), 125.60 (t, J = 44.9 Hz), 111.18 (s). GPC: Mn = 15.5 kDa, Mw = 60.5 kDa, PDI = 3.9.
PDSKK55. Black product. Yield: 74%. Calc. for C480H320N40O35S15: C, 74.98; H, 4.20; N, 7.29; O, 7.28; S, 6.25. Found: C, 67.39; H, 5.54; N, 7.24; S, 5.54.
PDNS. Yellow product. Yield: 70%. 1H-NMR (400 MHz, CDCl3) δ 8.80 (t, J = 9.7 Hz, 1H), 8.59 (d, J = 9.9 Hz, 2H), 7.96–7.70 (m, 7H), 7.69–7.48 (m, 4H), 7.36 (d, J = 4.1 Hz, 1H). 13C NMR (101 MHz, CDCl3) δ 175.21 (s), 159.44 (s), 147.99 (s), 144.35 (s), 143.38 (s), 137.30 (s), 135.29 (s), 133.84 (d, J = 27.4 Hz), 132.35 (s), 130.48 (d, J = 31.8 Hz), 130.02 (s), 129.60 (s), 129.16 (d, J = 31.3 Hz), 128.35 (d, J = 51.0 Hz), 128.09 (s), 128.09 (s), 127.26 (s), 125.94 (s), 124.38 (d, J = 37.8 Hz). GPC: Mn = 24.6 kDa, Mw = 91.0 kDa, PDI = 3.7.
PDNK. Brown product. Yield: 74%. 1H-NMR (400 MHz, CDCl3) δ 8.65 (s, 1H), 8.21 (s, 1H), 8.04–7.86 (m, 5H), 7.73 (t, J = 13.4 Hz, 7H), 7.57 (s, 2H), 7.38 (s, 1H). 13C NMR (101 MHz, CDCl3) δ 171.58–171.03 (m), 155.08 (s), 144.15 (s), 142.17 (s), 139.58 (s), 136.54–136.11 (m), 135.89 (s), 131.50 (s), 130.03 (s), 128.39 (s), 126.68 (d, J = 40.2 Hz), 126.44 (s), 125.95 (s), 125.45 (s), 125.02 (s), 124.42 (d, J = 28.3 Hz), 124.28–124.01 (m), 123.20 (s), 121.96 (t, J = 34.6 Hz), 121.34 (s), 121.09–120.82 (m). GPC: Mn = 16.5 kDa, Mw = 59.4 kDa PDI = 3.6.
Results and discussion
Monomers
Monomers synthesis. For designing “non-ether bond” poly(phthalazinone ether) synthesized by classical nucleophilic aromatic substitution reactions, the monomer M1 was prepared by Stille coupling as showed in Scheme 2. The reactive monomer 2,5-bis(tributylstannyl) thiophene 1 was synthesized as literature reported.25 During Stille coupling reaction, the target monomer M1 was obviously precipitated out from the reactive medium NMP at 110 °C. Unconverted reactants and some by-products, detected by HPLC-MS in NMP (Fig. S11†), were removed by successively treated with hot DMAC, NMP and water, and then Soxhlet extracted with ethanol for 24 h. Unfortunately, the sufficient characterizations of M1 (1H-NMR, 13C-NMR and MALDI-TOF) were hard to carry out owing to its insolubility in organic solvent. Due to large molecular conjugated structure, melting point (measured by DSC) of M1 (Fig. S10†) was not detected from 30 to 350 °C. And only elemental analysis and Fourier transform infrared spectrometry (Fig. S8†) were obtained.For the detailed characterizations of M1, its long-chain alkyl substituted derivative M2 was synthesized. As we expected, M2 has excellent solubility in organic solvent. Thus 1H-NMR, 13C-NMR (Fig. S1†) and MALDI-TOF (Fig. S7†) of M2 were easily measured. The 1H-NMR, 13C-NMR spectra of M2 obtained all displayed the expected signals with no discernible peaks corresponding to impurities. Thus the structure of M1 was confirmed by the unambiguous spectral characterization of M2. The optical electrochemical properties and quantum chemical calculations (B3LYP/6-31G**) by Gaussian 09 package were also investigated as follows.
Molecular orbital computation. For further understanding electronic structure of M1, quantum chemistry calculations by DFT (B3LYP/6-31G**) method were performed (the hexyl group replaced the dodecyl group). As shown in Fig. 2, due to the electron-withdrawing group such as CN and CO in phthalazinone group, the LUMO orbits of M2 are delocalized along phthalazinone–thiophene axis and mainly localized at the phthalazinone core. While its HOMO orbits are delocalized on the thiophene core because of electron-rich of thiophene unit. If M2 is excited, its electrons will transfer from the thiophene core to phthalazinone–thiophene core. The calculated HOMO–LUMO gap of M2 was found to be 3.64 eV. In addition, a couple of sensitive and reversible redox peaks were obtained from the cyclic voltammograms (CV) (details in Fig. S9†). The CV-calculated HOMO and LUMO were −5.87 eV and −2.79 eV, respectively. This indicates that M1 has the strong electron-withdrawing ability and can be used as acceptor to build conjugated polymers.
 |
| | Fig. 2 Calculated LUMO and HOMO orbits for M2 (the hexyl group replaced the dodecyl group). | |
Optical properties. UV/vis absorption spectra of M2 were studied in different solvent (Fig. 3). The UV/vis absorption of M2 showed slight bathochromic shift effect with the increase in the polarity of solvents. The maximum absorption wavelength of M2 in solution ranged from 345 nm to 353 nm, and its long wavelength of the absorption tails reached to 393 nm to 413 nm, respectively.
 |
| | Fig. 3 UV-vis absorption spectra for M2 in different solvent. | |
Fluorescence spectra of M2 in different solvent are shown in Fig. 4. M2 showed two emission peaks, and bathochromic shift effect was also observed with the increase in the polarity of solvents. Its second peak intensity increased with the polarity of solvents. In NMP, the emission peak became broadened by virtue of intermolecular charge transfer (ICT) effect of donor–acceptor structure (phthalazinone–thiophene). In addition, M2 exhibited a strong emissivity (Table 1) and was different from other oligothiophenes' and polythiophenes' weak-to-moderate emitters, due to the heavy atom effect of sulfur to enhance the intersystem. We suppose that intercrossed-excited-state (LE and CT) effect of M2 lead to the strong blue emissivity, which was confirmed by the DFT calculation below (Table 4) of the obvious twist angle between the phenyl, phthalazinone and thiophene units.26
 |
| | Fig. 4 Emession spectra of M2 in different solvent. | |
Table 1 Fluorescence property of M2
| Solvent |
Excitation wavelength (nm) |
Emission wavelength (nm) |
PLQY |
| Chloromethane |
345 |
413.2, 430.4 |
0.45 |
| Diethyl ether |
345 |
407.6, 426.6 |
0.54 |
| n-Hexane |
345 |
406.8, 426.0 |
0.52 |
| Methyl-2-pyrrolidinone |
345 |
420.4, 434.6 |
0.53 |
Polymer
Polymer synthesis. Ten polymers were then prepared by the classical nucleophilic aromatic substitution reactions between di-NH capped monomer M1 and various activated di-halogenated compounds with the yield being 70–79% (Table 2). A strong base (Cs2CO3) and strong polar, high boiling solvent (hexamethylphosphoric triamide: HMPA) were used in order to obtain high molecular polymers in our polymerization system. The number-average moleculars (Mn) of the obtained polymers were varying from 15.0 kDa to 24.6 kDa. Their polydispersity index (PDI), and N% (the N content of the polymers, measured by elemental analysis) are summarized in Table 2. The values of N% showed good agreement between calculated and measured. All resulting polymers had a much broader distribution of molecular weight, presumably due to monomer's (M1) poor solubility to lead to the rather high viscosity of the reaction mixture and make the heating and mixing of the reactants uneven.27 All polymers' solubility in organic solvents are shown in Table S1.† PDSK55 and PDSKK55 were insoluble in organic solvent due to the high content of linear rod-like structure (ketone groups) in the polymer's chain. PDK and PDKK, prepared from 4,4′-difluoro diphenylmethanone (monomer M4) or 4,4′-difluorobenzoyl benzene (monomer M5), respectively, were precipitated during polymerization. However, naphthalene series of polymers (PDNS and PDNK) had better solubility in organic solvent than phenyl series of polymers. This can be explained by the optimized molecular geometries obtained by DFT as shown in Table 4. The big twist angle θ3 in the main chain of naphthalene series of the polymers results in its excellent solubility in organic solvent.
Table 2 Synthetic data of copolymers
| Polymer |
Mn (kDa) |
PDI |
N% (cal/found) |
Yielda (%) |
| Isolated after precipitation and purification. |
| PDS |
24.4 |
3.8 |
7.73/7.74 |
73 |
| PDSK82 |
16.5 |
3.3 |
7.75/7.49 |
74 |
| PDSK55 |
— |
— |
7.69/6.54 |
79 |
| PDSKK82 |
15.5 |
3.9 |
7.53/8.47 |
79 |
| PDSKK55 |
— |
— |
7.29/7.24 |
74 |
| PDNS |
24.6 |
3.7 |
6.79/7.47 |
70 |
| PDNK |
16.5 |
3.6 |
6.16/6.54 |
74 |
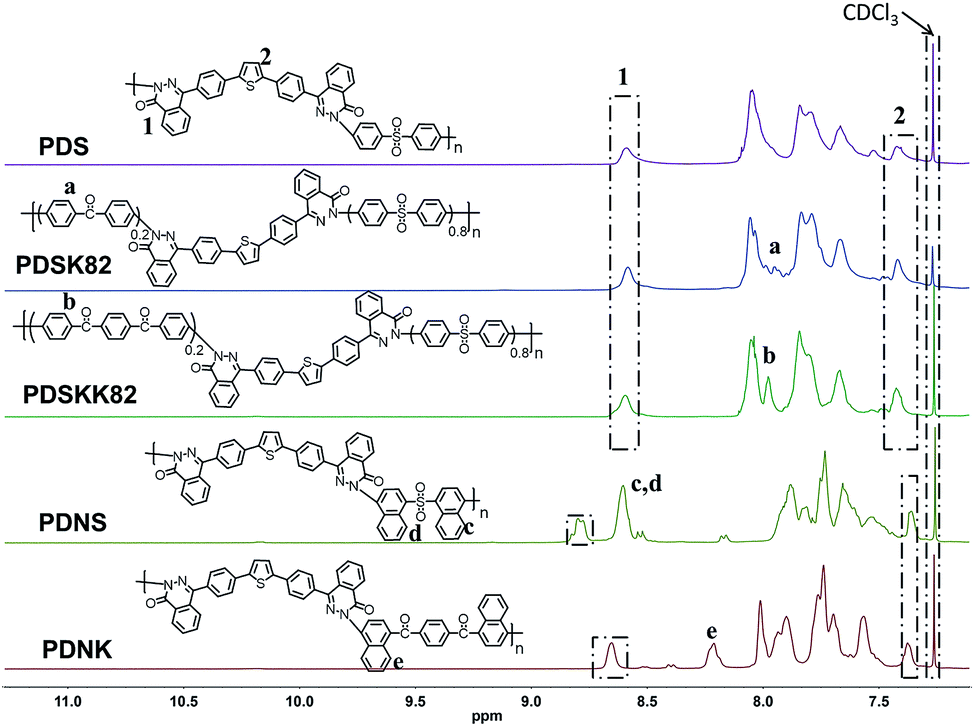
The structures of the resulting polymers were analyzed using 1H-NMR (Fig. 5) and 13C-NMR. The single peak splitting and shifting downfield at 8.59 ppm was attributed to the typical peri-position signal of the phthalazinone, which is always used as the reference signal to assign the other atom except PDNS. For PDNS, the chemical shift of the peri-position proton in the phthalazinone was shifted to lower field about 8.78–8.80 ppm due to the sulfone group and naphthalene. The chemical shift of the protons of the thiophene core appeared at about 7.40 ppm. The spectroscopic dates in Fig. 5 are in agreement with the proposed structures.
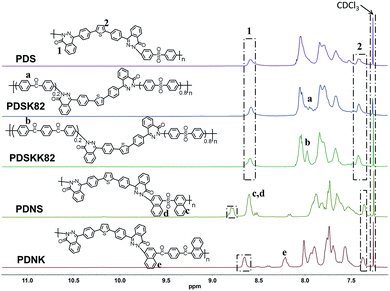 |
| | Fig. 5 1H-NMR spectra of polymers. | |
Furthermore, their 13C-NMR pictures are shown in Fig. 6. A detailed chemical shift assignment of our copolymer products are figured out. It is revealed that the PDNS and PDNK can be clearly distinguished from other polymers. For the PDSK82 and PDSKK82 copolymers, their different signals from PDS were attributed to M2 and M3 units, which is successfully in agreement with the copolymer structures as we designed.
 |
| | Fig. 6 13C-NMR spectra of polymers. | |
Other chemical shifts of 1H-NMR and 13C-NMR spectra were also found to be well in accord with the chemical structures of the polymers, as listed in Experimental sections.
The structure of the polymers obtained was also confirmed by Fourier transform infrared (FTIR) spectroscopy as depicted in Fig. 7. A asymmetric stretching vibration band at 1300 cm−1 to 1350 cm−1 and a symmetrical stretching vibration band at 1120 cm−1 to 1160 cm−1 from OSO stretch were observed for PDS, PDSK and PDSKK polymers containing sulfone groups, also for PDNS. The ketone groups of the polymers could be confirmed by C–C stretch on CO group at 1260 cm−1.
 |
| | Fig. 7 FTIR spectra of the different copolymers in powder. | |
Thermal properties. Thermal stability of all polymers was investigated by using thermogravimetric analysis (TGA) with a heating ramp rate of 20 °C min−1 under a N2 atmosphere (Fig. S12†). The 5% weigh loss temperatures (Td5%) of all soluble polymers (after purified) were above 489 °C after removing a small amount of residual water, HMPA and NMP below 250 °C, demonstrating the excellent thermal stability of these polymers. Due to the insolubility of PDSK55 and PDSKK55, they were not purified (only Soxhlet extracted by ethanol) in the result that the Td5% of PDSK55 and PDSKK55 was only 441 and 460 °C because of the residual low molecular weight oligomers. In addition, naphthalene series of polymers exhibited higher char yield (72%–75%) than phenyl series of polymers (63%–68%). The char yield of the investigated polymers in the nitrogen atmosphere was in the range of 63–75% at 800 °C, confirming their excellent thermal stability.Differential scanning (DSC) was implemented at a scan rate of 10 °C min−1 to investigate the phase transition of all polymers, as shown in Fig. S14† and Table 3. The glass transition temperatures (Tg) of these polymers were recorded in exceed 213 °C. Among the investigated polymers, PDS displayed the highest Tg value exceed 350 °C. The reasonable explains are that the strongest interaction of the polymer chains of PDS with the introduction of polar sulfone groups to hinder the movement of polymer chains. In addition, the Tg values of the detected polymers decreased gradually with the increasing content of ketone groups in the main chain of the polymers for the above reasons.28
Table 3 Thermal properties of copolymers
| Polymers |
Tg (°C) |
T5% (°C) |
Cy (%) |
| PDS |
>350 |
492 |
63 |
| PDSK82 |
326 |
495 |
67 |
| PDSK55 |
288 |
441 |
74 |
| PDSKK82 |
272 |
489 |
68 |
| PDSKK55 |
261 |
460 |
74 |
| PDNS |
267 |
496 |
72 |
| PDNK |
213 |
495 |
75 |
| M1 |
— |
403 |
33 |
Molecular orbital computation. To receive the information on the electronic states of the synthesized polymers, we performed the theoretical calculation using density-functional theory (DFT) at the B3LYP/6-31G(d,p)//B3LYP/6-31G(d,p) (Fig. 8). PDS and PDSK82 have better extension of the conjugated system through the polymer main chains than PDSKK82, PDNS and PDNK. Meanwhile, a big twist angles of di-ketone and naphthalene unit in the polymer backbone resulted in weak extension of the conjugated system. This leads to the limitation of the conjugation effects of PDSKK82, PDNS and PDNK into only one segment. Also, the LUMO electrons clouds of the polymers containing ketone groups were distributed at ketone groups, which are different from those of PDS and PDNS containing sulfone groups. That indicates that N–H side in phthalazinone unit has excellent conjugated effect.
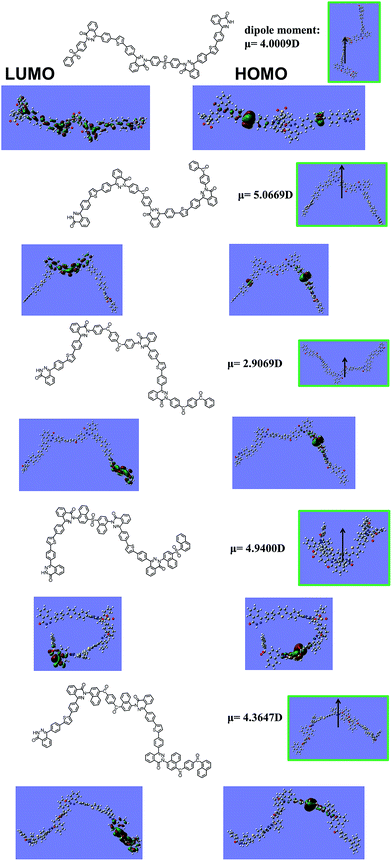 |
| | Fig. 8 Calculated dipole moment, HOMO and LUMO orbitals of polymers. The black arrow shows the magnitude and direction of the dipole moment. | |
The dihedral angles (deg) along the conjugated backbone for the optimized molecular geometries are also shown in Table 4. The dihedral angles between benzene and thiophene rings (θ1) were 19–24°.Comparing with θ2 = 41–44°, the naphthalene linked with benzene rings increased steric hindrance. Similarly, θ3 increased with the increasing steric hindrance from naphthalene with benzene rings (phenyl series of polymers) to naphthalene with naphthalene rings (naphthalene series of polymers). Also the dipole moment of the polymers is listed out in Fig. 8. The influence on optical properties and electrochemical properties in more details would be discussed as follows.
Table 4 Dihedral angles (deg) along the conjugated backbone for the optimized molecular geometries obtained by DFT evaluated at B3LYP/6-31G(d,p) level
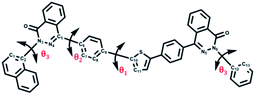
|
| Polymer |
θ1 (deg) |
θ2 (deg) |
θ3 (deg) |
| PDS |
22.7 |
43.4 |
29.3 |
| PDSK82 |
22.9 |
42.8 |
28.9 |
| PDSKK82 |
23.4 |
44.4 |
30.2 |
| PDNS |
19.2 |
41.1 |
60.0 |
| PDNK |
24.6 |
43.2 |
62.2 |
Optical properties. The UV-vis absorption and fluorescence spectra of all polymers were recorded in NMP at a concentration of 1 mg mL−1 and in drop-coated thin film. The UV-vis absorption and fluorescence spectra are shown in Fig. 9 and 10. All data are summarized in Table 5.
 |
| | Fig. 9 UV-vis absorption spectra for the polymers in NMP and thin film. | |
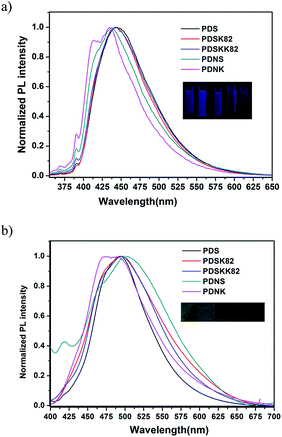 |
| | Fig. 10 Emission spectra of polymer in NMP (a) and thin film (b). | |
Table 5 Optical properties of polymers
| Polymers |
λmax (nm) |
λem (nm) |
Stokes shift (nm) |
PLQY |
| Solution |
Film |
Solution |
Film |
| PDS |
348, 354 |
402 |
448 |
496 |
94 |
0.20 |
| PDSK82 |
356 |
366 |
443 |
493 |
87 |
0.28 |
| PDSKK82 |
349 |
<350 |
443 |
496 |
94 |
0.17 |
| PDNS |
341, 350 |
365, 377 |
436 |
501 |
86 |
0.19 |
| PDNK |
346 |
364 |
434 |
495 |
88 |
0.04 |
The maximum absorption wavelength of PDS in solution was 348 nm (354 nm shoulder peak), which is 8 nm blue-shifted relative to PDSK82 (356 nm) and 1 nm blue-shifted relative to PDSKK82 (349 nm). Their dihedral angles had no obvious change. So the blue shift of absorption for PDS could be ascribed to weaker electron-withdrawing property of ketone group in PDSK82 and PDSKK82. From the other side, the ketone group in PDSK82 and PDSKK82 has stronger electron-donating ability to DHPZ core. This phenomenon also appeared for PDNS and PDNK. Moreover, due to the big dihedral angle of two naphthalene groups (θ3 in Table 4), the maximum absorption wavelength of PDS and PDSK82/KK82 (phenyl series) appeared at longer wavelength than PDNS and PDNK (naphthalene series). The maximum absorption peaks of the most polymers exhibited red shift effect than M2 (346 nm), indicating that the conjugated length of the polymers could be prolonged along the polymeric backbones due to cutting off the ether bond in the main chain of the polymers although prepared by classical nucleophilic displacement polycondensation reaction. In the solid state, the absorption spectra of all polymers were broadened; red shifted and exhibited a distinguishable structure. Especially, the absorption spectra for sulfone-containing polymers broadened and red shifted more than the polymers containing ketone and di-ketone segments. This is attributed to strong chain interaction of sulfone group than that of ketone group. Moreover, θ3 = 29–30° for phenyls series of polymers was smaller than θ3 = 60–62° for naphthalene series of polymers. Phenyl series of polymers like PDS have stronger intermolecular interaction than naphthalene series of polymers like PDNS. So the maximum absorption wavelength of PDS in film state was 402 nm, longer than that of PDNS (365 nm, 377 nm). All these above observations are usually associated with possible charge transport applications.
Generally, oligo-, polythiophenes and heteroaromatic polymers are weak-to-moderate emitters owing to the heavy atom effect of sulfur enhancing the intersystem crossing29 and the heterocyclic compounds with nitrogen heteroatoms spin-forbidding n–π*. As a surprise, herein we observed that PDSK82 (PLQY = 28%) containing fused thiophene ring and nitrogen-heterocyclic structure exhibited a strong photoluminescence, higher than PDS (PLQY = 20%) and PDSKK82 (PLQY = 17%). We speculate that PDSK82 containing ketone group has different localization manners between LUMO and HOMO than PDS and PDSKK82 in the result of better emission originated from the intermolecular charge transfer (ICT) states.26,29 However, PDSKK82 and PDNK containing di-ketone group exhibited weak emissive. The reason can be found that the two ketone groups interrupt their conjugated structure in the main chain to hinder the electron transport along the polymer backbone in Fig. 8. It is worth noting that due to the strong torsion between phenyl–phthalazinone core and naphthalene in M6 and M7, phenyl series of polymers show longer wavelength emission than naphthalene series of polymers in NMP.
Electrochemical properties. The electrochemical properties of the synthesized polymer are summarized in Table 6. The frontier molecular orbits determined by CV (Fig. S14†) is compared to the predict energies using DFT with a 6-31G** basis set as shown in Fig. 11. M2 showed two reversible oxidation processes and one irreversible redox processes during negative scan. Its energy levels were estimated to be −2.79 eV for the LUMO and −5.87 eV for the HOMO. And its energy band was 3.08 eV. All polymers derived from M2 exhibited a narrower electrical energy band than M2, indicating di-NH end-caped monomer could prolong conjugated length in the polymeric backbones. In addition, weak CT effect of sulfone and ketone groups resulted in the broad electrical energy band of all polymers than most conjugate D–A polymers.30 PDSK82 showed the most narrow optical energy band among these polymers. The reasons could be explained by DFT calculation in Fig. 8. It has been proved that a molecule having different localization manners between the frontier orbits often exhibits the broad emission in the long wavelength region originated from the intramolecular charge transfer (ICT) states. The optical energy band of five soluble polymers were varies from 2.73 eV to 2.88 eV.
Table 6 Electrochemical data of polymers
| Polymer |
LUMO (eV) |
HOMO (eV) |
Eg (eV) |
| Calculated by cyclic voltammograms. Calculated by Gaussian 09. Calculated by UV-vis absorption spectra. |
| PDS |
−2.75a |
−2.11b |
−5.51a |
−5.60b |
2.76a |
2.82c |
3.49b |
| PDSK82 |
−2.64 |
−2.05 |
−5.35 |
−5.55 |
2.71 |
2.73 |
3.50 |
| PDSKK82 |
−2.68 |
−2.23 |
−5.36 |
−5.59 |
2.68 |
2.86 |
3.36 |
| PDNS |
−2.80 |
−2.09 |
−5.16 |
−5.59 |
2.36 |
2.75 |
3.50 |
| PDNK |
−2.73 |
−2.32 |
−5.27 |
−5.55 |
2.54 |
2.88 |
3.23 |
 |
| | Fig. 11 Frontier molecular orbital energy levels as predicted by DFT calculation with a 6-31g** basis set and CV. | |
As showed in Fig. 11, the sulfone-containing polymer has deeper LUMO level than the ketone-containing analogue because of the strong electron-withdrawing ability of sulfone group. For instance, −2.75 eV for the LUMO of PDS was deeper than −2.64 eV (PDSK82) and −2.68 eV (PDSKK82). The electron energy band of naphthalene series of polymers like PDNS (2.36 eV) and PDNK (2.45 eV) was smaller than that of phenyl series of polymers like PDS (2.76 eV), PDSK82 (2.71 eV) and PDSKK82 (2.67 eV). However, PDNS and PDNK containing naphthalene segments showed the big dihedral angle in the polymer backbone, resulting in weak extension of the conjugated system. The CV measurement exhibited the narrower band gap for PDNS and PDNK than that value for other polymers with better co-planarity. As the naphthalene group has better electron-donating ability than phenyl, and the sulfone group has electron-drawing ability than ketone group, a strong donor–acceptor system also led to narrower band gap. We deduce that the latter plays a major factor under such circumstances.
From the onset of oxidation and reduction peaks the HOMO and LUMO values as well as the electrochemical band gaps were calculated. As shown in Table 6, the electrochemical band gap values calculated by CV were similar to the values predicted by DFT. The HOMO energy levels of most polymers were lower than their air oxidation (ca. −5.27 eV) indicating good air stability and a higher Voc in PSCs.31 Moreover, due to the low HOMO energy level of the polymers, M1 could be a potential acceptor to build conjugated polymers for solar cells.
Physical properties. X-ray diffraction (XRD) measurement was used to investigate the physical properties of the polymers in thin films. Fig. 12 shows the XRD patterns of the five polymers casted from NMP. Obviously, no sensitive peaks were found at the region from 5 to 60°, indicating that they possess some amorphous features without crystalline domains. Only a widely insensitive diffraction peaks (17°–30°) for π–π stacking of PDS, PDSK82 and PDNK were distinguished. Heeger and co-workers have put forth that self-assembly could be driven by the orientation and strength of the molecular dipole moment. As pointed out in Fig. 8, we can therefore hypothesize that the change of the molecular dipole moment results in a different intermolecular packing manner. Furthermore, it has been demonstrated experimentally and computationally that PDSK82 (dipole moment, μ = 5.0669 D) are more advantageous to the π−π stacking than PDSKK82 (dipole moment, μ = 2.9069 D). The different intensity of diffraction peaks (17°–30°) of PDSK82 and PDSKK82 can clearly be visible in Fig. 12. However, M6 with high dipole moment (μ = 4.0094 D) had no obvious widely insensitive diffraction peaks. That can be attributed to highly twisted backbone of PDNS (Fig. 8).32 In addition, the non-crystalline feature of polymers shows they could be favorable for their PLED applications.33
 |
| | Fig. 12 The XRD spectra of polymer films. | |
Conclusions
In summary, we have designed, synthesized, and characterized a new di-NH capped monomer and its polymers prepared by classical nucleophilic aromatic polymerization. Compared with commercial PEEK, poly(aryl ethers)s and traditional poly(phthalazinone ether)s synthesized by the same polymerization method, these non-ether bond polymers show remarkable photoelectric properties. To our knowledge, this is the first report that the polymers containing phthalazinone–thiophene structure derived from classical nucleophilic aromatic polymerization to apply in the optoelectronic polymer materials. The polymers exhibit highly thermostability and blue-fluorescence property. The optical band gap and HOMO energy levels are varying from 2.36 eV to 2.76 eV and −5.16 eV to −5.51 eV, respectively. The effect of ketone/di-ketone, sulfone group, phenyl and naphthalene groups in the main chain of the resulting polymers on the optical properties were investigated in details. On the basis of our results, fused di-NH monomer and classical nucleophilic displacement reaction method can narrower the band gap and adjust the wavelength of the luminescence. Further studies on the polymers based on di-NH capped monomer by classical nucleophilic aromatic polymerization are under way.
Acknowledgements
The present research was financially supported by National Natural Science Foundation of China (no. 21074017 and 51273029). The authors acknowledge the High Performance Computing Centre of Dalian University of Technology for providing computational resources which have contributed to the research results.
Notes and references
-
(a) G. Yu, J. Gao, J. C. Hummelen, F. Wudl and A. J. Heeger, Science, 1995, 270, 1789 CAS;
(b) X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 38, 1832–1908 CrossRef CAS PubMed;
(c) J. You, L. Dou, Z. Hong, G. Li and Y. Yang, Prog. Polym. Sci., 2013, 38, 1909–1928 CrossRef CAS PubMed;
(d) A. Sui, X. Shi, H. Tian, Y. Geng and F. Wang, Polym. Chem., 2014, 5, 7072–7080 RSC;
(e) C. Gao, L. Wang and X. Li, Polym. Chem., 2014, 5, 5200–5210 RSC.
-
(a) J. Hassan, M. Sévignon, C. Gozzi, E. Schulz and M. Lemaire, Chem. Rev., 2002, 102, 1359–1470 CrossRef CAS PubMed;
(b) P. Espinet and A. M. Echavarren, Angew. Chem., Int. Ed., 2004, 43, 4704–4734 CAS;
(c) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef CAS PubMed;
(d) B. Liu, B. Qiu, X. Chen, L. Xiao, Y. Li, Y. He and Y. Zou, Polym. Chem., 2014, 5, 5002–5008 RSC;
(e) G. Zhang, J. Guo, J. Zhang, P. Li, J. Ma, X. Wang and L. Qiu, Polym. Chem., 2015, 6, 418–425 RSC;
(f) L. Wang, D. Cai, Z. Yin, C. Tang, S. C. Chen and Q. Zheng, Polym. Chem., 2014, 5, 6847–6856 RSC.
- J. Wang, Y. Gao, A. R. Hlil and A. S. Hay, Macromolecules, 2008, 41, 298–300 CrossRef CAS.
- N. Berard and A. S. Hay, Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.), 1993, 34, 148–149 CAS.
-
(a) S. Yoshida and A. S. Hay, Macromolecules, 1995, 28, 2579–2581 CrossRef CAS;
(b) S. Yoshida and A. S. Hay, Macromolecules, 1997, 30, 2254–2261 CrossRef CAS.
- X. Li and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2007, 45, 975–979 CrossRef CAS.
- S. J. Wang, Y. Z. Meng, A. R. Hlil and A. S. Hay, Macromolecules, 2004, 37, 60–65 CrossRef CAS.
-
(a) L. Cheng, X. G. Jian and S. Z. Mao, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 3489–3496 CrossRef CAS;
(b) L. Cheng and X. G. Jian, J. Appl. Polym. Sci., 2004, 92, 1516–1520 CrossRef CAS.
-
(a) J. Y. Wang, X. G. Jian and S. D. Xiao, Chin. Chem. Lett., 2001, 12, 593–594 CAS;
(b) J. Y. Wang, G. X. Liao, C. Liu and X. G. Jian, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 6089–6097 CrossRef CAS.
- Y. R. Gao, J. Y. Wang, C. Liu and X. G. Jian, Chin. Chem. Lett., 2006, 17, 140–142 CAS.
- X. Li, Y. Gao, Q. Long and A. S. Hay, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1761–1770 CrossRef CAS.
-
(a) S. Xiao, J. Wang, K. Jin, X. Jian and Q. Peng, Polymer, 2003, 44, 7369–7376 CrossRef CAS PubMed;
(b) Y. Song, J. Wang, G. Li, Q. Sun, X. Jian, J. Teng and H. Zhang, Polymer, 2008, 49, 724–731 CrossRef CAS PubMed;
(c) L. M. Dong, G. X. Liao, C. Liu, S. S. Yang and X. G. Jian, Surf.Rev. Lett., 2008, 15, 705–709 CrossRef CAS.
-
(a) Y. Dai, X. Jian, S. Zhang and M. D. Guiver, J. Membr. Sci., 2001, 188, 195–203 CrossRef CAS;
(b) P. Y. Qin, X. J. Hong, M. N. Karim, T. Shintani, J. D. Li and C. X. Chen, Langmuir, 2013, 29, 4167–4175 CrossRef CAS PubMed.
- W. Qi, C. Lu, P. Chen, L. Han, Q. Yu and R. Xu, Mater. Lett., 2012, 66, 239–241 CrossRef CAS PubMed.
- D. Xing, S. Zhang, C. Yin, B. Zhang and X. Jian, J. Membr. Sci., 2010, 354, 68–73 CrossRef CAS PubMed.
- J. Pan, K. Li, S. Chuayprakong, T. Hsu and Q. Wang, ACS Appl. Mater. Interfaces, 2010, 2, 1286–1289 CAS.
-
(a) X. Ma, C. Zhang, G. Xiao, D. Yan and G. Sun, J. Polym. Sci., Part A: Polym. Chem., 2008, 46, 1758–1769 CrossRef CAS;
(b) H. G. Chen, S. J. Wang, M. Xiao and Y. Z. Meng, J. Power Sources, 2007, 165, 16–23 CrossRef CAS PubMed.
-
(a) H. Zhang and B. Tieke, Polym. Chem., 2014, 5, 6391–6406 RSC;
(b) Z. G. Zhang and J. Wang, J. Mater. Chem., 2012, 22, 4178–4187 RSC.
- V. A. Naumov, S. Samdal, A. V. Naumov, S. Gundersen and H. V. Volden, Russ. J. Gen. Chem., 2005, 75, 1956–1961 CrossRef CAS PubMed.
-
(a) A. Pron and M. Leclerc, Prog. Polym. Sci., 2013, 8, 1815–1831 CrossRef PubMed;
(b) X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- T. C. Chao, Y. T. Lin, C. Y. Yang, T. S. Hung, H. C. Chou, C. C. Wu and K. T. Wong, Adv. Mater., 2005, 17, 992–996 CrossRef CAS.
-
(a) T. Yokozawa and A. Yokoyama, Chem. Rev., 2009, 109, 5595–5619 CrossRef CAS PubMed;
(b) P. David, B. Jan, G. T. Hendrik, W. W. Gary, D. Ernst-Ulrich, O. Edgar and R. Klaus, Polymers, High-Temperature. Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2012, DOI:10.1002/14356007.a21_449.pub3.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- C. A. Heller, R. A. Henry, B. A. McLaughlin and D. E. Bliss, J. Chem. Eng. Data, 1974, 19, 214–219 CrossRef CAS.
- X. Guo and D. W. Mark, Org. Lett., 2008, 10, 5333–5336 CrossRef CAS PubMed.
- W. Li, D. Liu, F. Shen, D. Ma, Z. Wang, T. Feng and Y. Ma, Adv. Funct. Mater., 2012, 22, 2797–2803 CrossRef CAS.
-
(a) L. Fang, Y. Zhou, Y. X. Yao, Y. Lee, W. Y. Diao, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. N. Bao, Chem. Mater., 2013, 25, 4874 CrossRef CAS;
(b) Z. Zhao, F. Zhang, Y. Hu, Z. Wang, B. Leng, X. Gao, C. Di and D. Zhu, ACS Macro Lett., 2014, 3, 1174–1177 CrossRef CAS.
- G. P. Yu, C. Liu, J. Y. Wang, T. S. Gu and X. G. Jian, Polym. Degrad. Stab., 2009, 94, 1053–1060 CrossRef CAS PubMed.
-
(a) I. F. Perepichka, D. F. Perepichka, H. Meng and F. Wudl, Adv. Mater., 2005, 17, 2281 CrossRef CAS;
(b) J. A. Schneider, A. Dadvand, W. Wen and D. F. Perepichka, Macromolecules, 2013, 46, 9231–9239 CrossRef CAS.
- X. Wang, L. Zhao, S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, Macromolecules, 2014, 47, 2907–2914 CrossRef CAS.
-
(a) B. C. Thompson, Y. G. Kim and J. R. Reynolds, Macromolecules, 2005, 38, 5359–5362 CrossRef CAS;
(b) L. D. M. Simenon, M. M. J. Brown, A. R. Einerhand and R. E. F. De, Synth. Met., 1997, 87, 53–59 CrossRef.
-
(a) J. S. Wu, J. F. Jheng, J. Y. Chang, Y. Y. Lai, K. Y. Wu, C. L. Wang and C. S. Hsu, Polym. Chem., 2014, 5, 6472–6479 RSC;
(b) J. F. Jheng, Y. Y. Lai, J. S. Wu, Y. H. Chao, C. L. Wang and C. S. Hsu, Adv. Mater., 2013, 25, 2445–2451 CrossRef CAS PubMed;
(c) C. J. Takacs, Y. Sun, G. C. Welch, L. A. Perez, X. Liu, W. Wen and A. J. Heeger, J. Am. Chem. Soc., 2012, 134, 16597–16606 CrossRef CAS PubMed.
-
(a) S. Zhang, L. Ye, W. Zhao, D. Liu, H. Yao and J. Hou, Macromolecules, 2014, 47, 4653–4659 CrossRef CAS;
(b) C. W. Ge, C. Y. Mei, J. Ling, F. G. Zhao, H. J. Li, L. Liang and W. S. Li, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 2356–2366 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR and 13C-NMR spectrum of M2 and polymers, MALDI-TOF, IR, DSC, CV and HPLC-MS spectrum of M2, DSC and CV curves of polymers, solubility table of monomers and copolymers in organic solvent. See DOI: 10.1039/c5ra03771a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.