
Visible light driven photocatalytic elimination of organic- and microbial pollution by rutile-phase titanium dioxides: new insights on the dynamic relationship between morpho-structural parameters and photocatalytic performance
Abstract
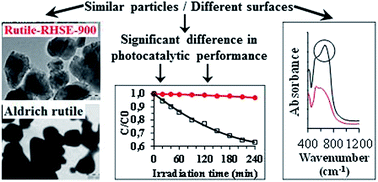
The characteristic properties and the resulted photocatalytic efficiencies of rutile-phase titanium dioxides were investigated in the present study. A series of rutile with different primary particle sizes (5.2–290 nm) were produced by a sol–gel method followed by calcination and were characterized by XRD, DRS, TEM, XPS, EPR, IR and N2 adsorption. Their photocatalytic efficiencies were determined in the decomposition of phenol, and in the inactivation of E. coli bacteria under visible light irradiation. The results were compared with the photocatalytic performance of commercial Aldrich rutile and Aeroxide P25 powders. Of the non-commercial products, the TiO2 with the smallest particle size displayed the highest efficiency, while the surface-normalized photocatalytic performance was significantly higher for the larger rutile particles. This can be explained by the red shift of light absorption at higher calcination temperatures. Although Aldrich rutile and the corresponding laboratory-made photocatalyst exhibited similar structural features (e.g. particle size, specific surface area, morphology and light absorption), the latter proved to be less efficient despite its Ti3+ content (while Aldrich rutile contains only Ti4+). The main reason for the much higher photocatalytic performance was the presence of Ti–O–O– entities on the surface of Aldrich rutile. On the basis of these results, in the case of rutile-phase titanium dioxide, the presence of Ti–O–O– entities was more beneficial, than the presence of Ti3+ and low-binding-energy oxygen (which indicates defects) in relation with the photocatalytic performance under visible light irradiation.
 Please wait while we load your content...
Please wait while we load your content...

