
Platinum(ii) polymetallayne-based phosphorescent polymers with enhanced triplet energy-transfer: synthesis, photophysical, electrochemistry, and electrophosphorescent investigation†
Abstract
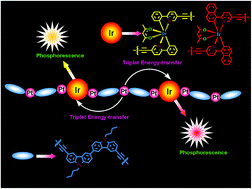
Two series of new phosphorescent copolymers with bicarbazole-based platinum(II) polymetallayne backbones have been successfully prepared through Sonogashira cross-coupling with different IrIII ppy-type (ppy = 2-phenylpyridine anion) complexes as phosphorescent centers. The photophysical investigations not only indicate a highly efficient triplet energy-transfer process from the polymetallayne segments to the phosphorescent units in the polymer solution, but also figure out the structure–property relationship between the triplet energy-transfer process and the energy-levels of different excited states. In addition, the phosphorescent copolymers can produce yellow-emitting phosphorescent OLEDs (PHOLEDs) with high EL efficiencies and a current efficiency (ηL) of 11.49 cd A−1, an external quantum efficiency (ηext) of 4.38%, a power efficiency (ηP) of 3.78 lm W−1, and red-emitting PHOLEDs with a ηL of 5.86 cd A−1, ηext of 10.1%, and a ηP of 2.29 lm W−1, representing very decent electroluminescent performances achieved by the phosphorescent copolymers. Herein, this work not only furnishes very important clues for further polishing of this category novel phosphorescent polymer, but also provides a new approach to the design and synthesis of highly efficient phosphorescent copolymers.
 Please wait while we load your content...
Please wait while we load your content...

