DOI:
10.1039/C5RA03693C
(Communication)
RSC Adv., 2015,
5, 34040-34046
First total synthesis of cryptomoscatone F1†
Received
2nd March 2015
, Accepted 31st March 2015
First published on 1st April 2015
Abstract
The total synthesis of cryptomoscatone F1 has been accomplished for the first time. The strategy involves chelation-controlled allylation, the addition of alkylzinc to an aldehyde and olefin cross-metathesis reactions as key steps. a new route to a vinyl lactone was explored.
Styryl lactones such as cryptomoscatones D1,1 D2, E1, E2 (cryptofolione), E3, and F1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2 (Fig. 1) were isolated from branch and stem bark of Cryptocarya mandiocanna, and C. moschata Lauraceae. Their structures were established by spectroscopic methods. C moschata Nees is an arboreal species that is widespread in the Atlantic forests of Brazil. It is recognised as an important food source for primates such as Brachyteles arachnoids (E. Geoffroy, 1806). Cryptocarya is one of the largest pantropical genera in Lauraceae. The genus includes about 350 species, and lactones isolated from these species have been shown to exhibit several biological activities.3 Cryptomoscatone D2 possesses high dose-dependent and time-dependent cytotoxicity in HeLa, SiHa, C33A, and MRC-5 cell lines.4 Cryptofolione showed activity towards Trypanosoma cruzi trypomastigotes, reducing their number by 77% at 250 μg mL−1.5
2 (Fig. 1) were isolated from branch and stem bark of Cryptocarya mandiocanna, and C. moschata Lauraceae. Their structures were established by spectroscopic methods. C moschata Nees is an arboreal species that is widespread in the Atlantic forests of Brazil. It is recognised as an important food source for primates such as Brachyteles arachnoids (E. Geoffroy, 1806). Cryptocarya is one of the largest pantropical genera in Lauraceae. The genus includes about 350 species, and lactones isolated from these species have been shown to exhibit several biological activities.3 Cryptomoscatone D2 possesses high dose-dependent and time-dependent cytotoxicity in HeLa, SiHa, C33A, and MRC-5 cell lines.4 Cryptofolione showed activity towards Trypanosoma cruzi trypomastigotes, reducing their number by 77% at 250 μg mL−1.5
Thus, interesting biological activities of cryptomoscatones, along with limited availability from natural sources, impelled us to undertake a total synthesis of cryptomoscatone F1. We have reported the synthesis of cryptomoscatone D26a and cryptofolione.7 Recently, similar cryptomoscatones6b,c,d have attracted the attention of other synthetic research groups. Now, we describe herein the first total synthesis of cryptomoscatone F1 (6). It was proposed a threo relationship between OH-4′ and OH-6′ and erythro relationship between OH-6′ and OH-8′.
Our synthetic plan toward 6 is summarized in Scheme 1. We envisaged that F1 could be derived from the triol 7 and the vinyl lactone 8 via olefin cross-metathesis (CM). While vinyl lactone 8 could be prepared from commercially available (R)-2,3-O-cyclohexylidene glyceraldehyde8 by a new synthetic route.
 |
| | Scheme 1 Retrosynthetic analysis for 6. | |
 |
| | Fig. 1 Natural cryptomoscatones 1–6. | |
Results and discussion
The synthesis of triol 7, depicted in Scheme 2, started with protection of 12 [(S)-enantiomer] (prepared as reported for its known R-enantiomer)9 as its benzyl ether using benzyl trichloroacetimidate and CSA in CH2Cl2 to give 14 in 84% yield. To establish the second stereogenic center with the required stereochemistry, it was thought worthwhile to adopt a chelation-controlled stereoselective allylation reaction occurring through 1,3-induction. Accordingly, oxidative cleavage of the olefin in compound 14 under Jin's one-pot conditions10 using OsO4–NaIO4 and 2,6-lutidine in dioxane–water (3:1) furnished the corresponding aldehyde, which was immediately treated with allyltrimethylsilane in the presence of MgBr2·OEt211 in CH2Cl2 at 0 °C to afford homoallyl alcohol 15 in 86% yield with a diastereomeric ratio of 20:1. The benzyl group was deprotected with Li/naphthalene in dry THF to give diol 16, which was subsequently transformed into isopropylidene derivative 17 with dimethoxypropane and a catalytic amount of PPTS in 90% yield. The TBDPS group in compound 17 was removed with TBAF in THF and the anti geometry was assigned to 1,3-diol group in compound 11 by Rychnovsky method.12 In the 13C NMR spectrum of 11, the resonance arising from the acetonide methyl groups appeared at δ = 24.64 and 24.75 ppm and that of the quaternary carbon atom at δ = 100.29 ppm, indicating a 1,3-anti relationship.
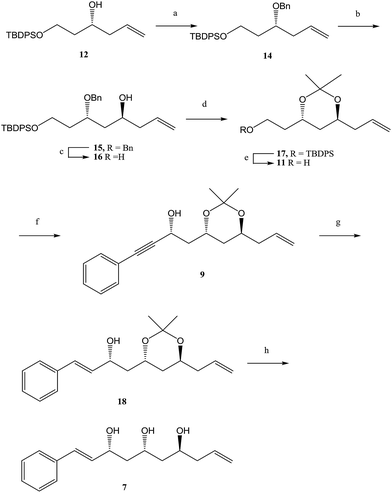 |
| | Scheme 2 Synthesis of triol 7. reagents (a) BNOC(NH)CCl3, CSA, CH2Cl2, rt, 12 h, 84%; (b) (i) OsO4, NaIO4, 2,6-lutidine, 1,4-dioxane/H2O, rt, 2 h; (ii) allylSiMe3, MgBr2·Et2O, CH2Cl2, 0 °C, 12 h, 76% (over 2 steps); (c) Li/naphthalene, dry THF, 1 h, −10 °C, 85%; (d) 2,2-DMP, PPTS, 0 °C rt, 10 h, 94%; (e) TBAF, THF, 0 °C, 3 h, 90%; (f) IBX, AcCN, reflux, 1 h (ii) phenylacetylene, Et2Zn, (S)-BINOL, Ti(OiPr)4, toluene, dry CH2Cl2, 6 h, 74% (over 2 steps); (g) Red-Al, THF, 0 °C, 3 h, 93%; (h) CuCl2·2H2O, AcCN, 0 °C, 1 h, 90%. | |
After confirming the stereocenters, compound 11 on oxidation with IBX in CH3CN gave an aldehyde. Next, we planned to introduce C8′ chiral center by asymmetric alkynylzinc addition reaction to aldehyde.13 Thus, addition of phenylacetylene to an aldehyde in the presence of diethylzinc, (S)-BINOL, and Ti(OiPr)4 generated chiral propargyl alcohol with dr 95:5.14
Stereochemical assignment at the newly created hydroxy bearing center in 9 (C8′-OH in target molecule) was established as R by preparing R and S-mandelic esters. A reduction of the triple bond in compound 9 to the corresponding double bond was accomplished using Red-Al to give 18 in 92% yield. The acetonide group was removed upon treatment with CuCl2·2H2O in CH3CN at 0 °C to afford triol compound 7 in 90% yield.
The synthesis of vinyl lactone, other synthon was initiated from (R)-2,3-O-isopropylidene glyceraldehyde, which was subjected to 1,2-chelation-controlled allylation15 in the presence of MgBr2·(OEt)2, allyltributyltin to afford syn homoallylic alcohol 13 in 86% yield and dr 97:3.16 The diastereomeric ratio was determined by chiral HPLC column in the next step after conversion of homoallylic alcohol 13 into cinnamoyl ester. Thus, esterification of homoallylic alcohol 13 with cinnamoyl chloride furnished the corresponding ester, which was subjected to ring-closing metathesis (RCM) using Grubbs Ist generation catalyst to afford unsaturated lactone. The cyclohexylidene protecting group was easily removed with 80% TFA in CH2Cl2 to obtain diol, which on treatment with PPh3–imidazole–iodine17 in refluxing toluene converted to the terminal olefin affording vinyl lactone (8) (Scheme 3).
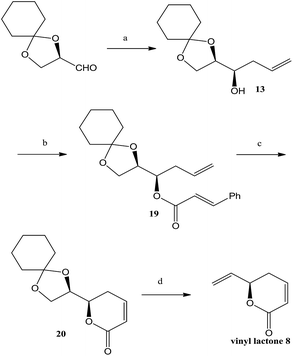 |
| | Scheme 3 Synthesis of vinyl lactone 8. Reagents: (a) allyltributyltin, MgBr2·Et2O, CH2Cl2, −78 °C to −20 °C, 16 h, 86%; (b) (E)-cinnamoyl chloride, Et3N, DMAP, CH2Cl2, 0 °C, 12 h, 85%; (c) G-I catalyst, dry CH2Cl2, reflux, 24 h, 92%; (d) (i) 80% TFA, CH2Cl2, 0 °C, 2 h, (ii) PPh3, Im, I2, toluene, reflux, 3 h, 68% (over 2 steps). | |
Having both fragments 7 and 8 in hand, we proceeded for olefin cross-metathesis reaction. Thus, triol 7 was subjected to cross-metathesis coupling with vinyl lactone 8 in the presence of Grubbs' second generation catalyst18 in refluxing CH2Cl2 for 1 h to afford the target, cryptomoscatone F1 in 89% yield (Scheme 4). The 1H and 13C NMR of our synthetic 6 are in full agreement with the reported spectra of natural product. NOESY experiment was also performed in order to confirm the E configuration of the double bonds in compound 6 (Fig. 2).
 |
| | Scheme 4 Synthesis of cryptomoscatone F1. Reagents (a) G-II catalyst, CH2Cl2, reflux, 1 h, 89%. | |
 |
| | Fig. 2 NOESY characteristic NOE correlations of compound 6. | |
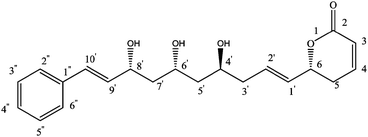
Determining the E-configuration of C1′![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2′ and C9′C10′ double bonds based on 1H NMR (500 MHz, CDCl3) data and assignments were made with the aid of NOESY experiments (Fig. 3). The medium NOE correlation observed between C1′H/C5H, C1′H/C3′H determining the E-configuration of C1′C2′ double bond. The medium NOE correlation observed between C9′H/C7′H, and strong NOE correlation between C9′H/PhH, C10′H/PhH determining the E-configuration of C9′C10′ double bond (Fig. 2). Since the remaining double bond geometry was deduced by 1H–1H coupling constants (JH-3,H-4 = 9.9 Hz (Z)-configuration).
C2′ and C9′C10′ double bonds based on 1H NMR (500 MHz, CDCl3) data and assignments were made with the aid of NOESY experiments (Fig. 3). The medium NOE correlation observed between C1′H/C5H, C1′H/C3′H determining the E-configuration of C1′C2′ double bond. The medium NOE correlation observed between C9′H/C7′H, and strong NOE correlation between C9′H/PhH, C10′H/PhH determining the E-configuration of C9′C10′ double bond (Fig. 2). Since the remaining double bond geometry was deduced by 1H–1H coupling constants (JH-3,H-4 = 9.9 Hz (Z)-configuration).
This was further supported by 1H–1H coupling constants.
| JH-3,H-4 = 9.9 Hz (Z)-configuration, |
| JH-1′, H-2′ = 15.5 Hz (E)-configuration, |
| JH-9′, H-10′ = 15.8 Hz (E)-configuration |
 |
| | Fig. 3 NOESY spectrum showing the characteristic NOE correlations of compound 6. | |
Conclusions
In conclusion, we have accomplished the first total synthesis of cryptomoscatone F1 (6) in 13 steps with an overall yield of 11.57%.
Comparative data for the natural and synthetic compound was provided in Table 1.
Table 1 Comparative data of natural product2 and synthetic compound (6)
|
| 1H NMR |
|
13C NMR |
| H |
Natural product |
Synthetic product |
C |
Natural product |
Synthetic product |
| 3 |
6.00 (dt, J = 10, 1 Hz) |
6.03 (dt, J = 9.9, 1.6 Hz) |
2 |
164.1 |
164.2 |
| 4 |
6.80 (dt, J = 10, 4 Hz) |
6.91–6.85 m |
3 |
121.4 |
121.3 |
| 5 |
2.40 m |
2.47–2.39 m |
4 |
144.8 |
144.9 |
| 6 |
4.90 (br q, J = 6 Hz) |
4.93–4.86 m |
5 |
29.7 |
29.6 |
| 11 |
5.70 (dd, J = 16, 6 Hz) |
5.69 (dd, J = 15.5, 6.5 Hz) |
6 |
77.9 |
77.9 |
| 21 |
5.90 (dt, J = 16, 7 Hz) |
5.92–5.84 m |
11 |
129.7 |
129.5 |
| 31 |
2.30 (t, J = 7 Hz) |
2.35–2.22 m |
21 |
131.4 |
131.4 |
| 41 |
4.00 m |
4.08–4.01 m |
31 |
40.3 |
40.2 |
| 51 |
1.70 m |
1.92–1.82 m, 1H |
41 |
68.1 |
67.9 |
| 61 |
4.30 m |
4.33–4.24 m |
51 |
42.3 |
42.4 |
| 71 |
1.70 m |
1.75–1.60 m, 3H |
61 |
70.0 |
69.6 |
| 81 |
4.60 m |
4.62–4.56 m |
71 |
42.9 |
42.9 |
| 91 |
6.20 (dd, J = 16, 6 Hz) |
6.23 (dd, J = 15.8, 6.5 Hz) |
81 |
73.6 |
73.3 |
| 101 |
6.60 (d, J = 16 Hz) |
6.60 (d, J = 15.8 Hz) |
91 |
130.2 |
130.0 |
| Ph |
7.30 m |
7.41–7.22 m, 5H |
101 |
131.6 |
131.6 |
| |
|
|
111 |
136.5 |
136.5 |
| |
|
|
211/611 |
126.5 |
126.4 |
| |
|
|
311/511 |
128.6 |
128.5 |
| |
|
|
411 |
127.8 |
127.6 |
Experimental section
General
All reactions were performed under inert atmosphere. All glassware apparatus used for reactions are perfectly oven/flame dried. Anhydrous solvents were distilled prior to use: THF from Na and benzophenone; CH2Cl2, DMSO from CaH2; MeOH from Mg cake. Commercial reagents were used without purification. Column chromatography was carried out by using silica gel (60–120 mesh) unless otherwise mentioned. Analytical thin layer chromatography (TLC) was run on silica gel 60 F254 pre-coated plates (250 μm thickness). Optical rotations [α]D were measured on a polarimeter and given in 10−1 deg cm2 g−1. Infrared spectra were recorded in CHCl3/KBr (as mentioned) and reported in wave number (cm−1). Mass spectral data were obtained using MS (EI) ESI, HRMS mass spectrometers. High resolution mass spectra (HRMS) [ESI+] were obtained using either a TOF or a double focusing spectrometer. 1H NMR spectra were recorded at 300, 500 and 13C NMR spectra 75125 MHz in CDCl3 solution unless otherwise mentioned, chemical shifts are in ppm downfield from tetramethylsilane and coupling constants (J) are reported in hertz (Hz). The following abbreviations are used to designate signal multiplicity: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad.
(R)-1-((R)-1,4-Dioxaspiro[4.5]decan-2-yl)but-3-en-1-ol (13). To a solution of crude aldehyde (0.8 g, 4.7 mmol) in dry CH2Cl2 (8 mL) was added MgBr2·Et2O (1.46 g, 5.64 mmol). After stirring for 15 min and cooling to −78 °C allyl tributyl tin (1.6 mL, 5.17 mmol) was added; stirring was continued for 16 h while the temperature slowly rose to −20 °C. The mixture was poured into 10% aq HCl (10 mL). The water phase was extracted with CH2Cl2 (3 × 10 mL). The combined organic extracts were washed with sat. aq NaHCO3 (10 mL) and brine, dried, the solvent was evaporated and the residue purified by column chromatography (30% hexane/EtOAc) to give 13 (0.858 g, 86%) as a colorless liquid. [α]25D: +1.72 (c 1.2, CHCl3). IR (neat) νmax: 3435, 2935, 2860, 1640, 1163, 1100, 1044 cm−1. 1H NMR (300 MHz, CDCl3): δ 5.91–5.75 (m, 1H), 5.20–5.09 (m, 2H), 4.05–3.87 (m, 3H), 3.83–3.73 (m, 1H), 2.38–2.12 (m, 2H), 1.69–1.49 (m, 6H), 1.45–1.22 (m, 4H). 13C NMR (CDCl3, 125 MHz): δ 133.9, 118.1, 109.5, 77.6, 70.3, 64.7, 37.5, 36.1, 34.7, 25.0, 23.9, 23.7. HRMS (ESI) for C12H20O3Na [M + Na]+ found 235.1345 calcd 235.1340.
(R)-1-((R)-1,4-Dioxaspiro[4.5]decan-2-yl)but-3-en-1-yl cinnamate (19). To the solution of alcohol 13 (0.76 g, 3.58 mmol) was dissolved in 10 mL of CH2Cl2 and cooled to 0 °C and (0.895 mg, 5.37 mmol) of cinnamoyl chloride, (1 mL, 7.16 mmol) of Et3N, and (35 mg, 0.29 mmol) DMAP were added, warmed to room temperature and stirred overnight. The mixture was then poured into brine and extracted with CH2Cl2 (2 × 10 mL). The organic phases were washed with 1 M aq HCl (5 mL) and brine (8 mL), dried over Na2SO4. The solvent was removed under aspirator vacuum, and the crude product was purified by column chromatography silica gel (20% hexane/EtOAc), to obtain 0.999 g (85%) of 19. [α]25D: +8.09 (c 0.8, CHCl3). IR (neat) νmax: 3081, 2936, 2861, 1714, 1638, 1449, 1202, 1166, 1100, 767 cm−1. 1H NMR (500 MHz, CDCl3): 7.69 (d, J = 16.0 Hz, 1H), 7.55–7.50 (m, 2H), 7.41–7.36 (m, 3H), 6.44 (d, J = 16.0 Hz, 1H), 5.86–5.77 (m, 1H), 5.18–5.06 (m, 3H), 4.24–4.19 (m, 1H), 4.09–4.04 (m, 1H), 3.89–3.84 (m, 1H), 2.56–2.49 (m, 1H), 2.47–2.39 (m, 1H), 1.65–1.53 (m, 6H), 1.44–1.23 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 166.1, 145.2, 134.2, 131.1, 130.3, 128.8, 128, 118, 117.7, 110.1, 75.8, 73, 65.8, 36, 35.3, 34.8, 25.1, 23.9, 23.7. HRMS (ESI) for C21H26O4Na [M + Na]+ found 365.1728 calcd 365.1721.
(R)-6-((R)-1,4-Dioxaspiro[4.5]decan-2-yl)-5,6-dihydro-2H-pyran-2-one (20). Grubbs' first-generation catalyst (0.225 g, 0.27 mmol) was added to a solution of 19 (0.9 g, 2.74 mmol) in dry CH2Cl2 (100 mL) at reflux, over 24 h. After completion of the reaction (TLC), the solvent was removed under reduced pressure and the residue was purified by column chromatography (40% hexane/EtOAc) to give 20 (0.6 g, 92%) as a colorless liquid. [α]25D: +15.8 (c 1.3, CHCl3). IR (neat) νmax: 2935, 2860, 1738, 1247, 1099, 1054, 816 cm−1. 1H NMR (300 MHz, CDCl3): δ 6.96–6.86 (m, 1H), 6.01 (dd, J = 9.8, 1.7 Hz, 1H), 4.31–3.98 (m, 4H), 2.67–2.55 (m, 1H), 2.52–2.38 (m, 1H), 1.64–1.49 (m, 8H), 1.48–1.27 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 163, 144.9, 121.1, 110.4, 78, 75.6, 66.5, 36.4, 34.4, 26.2, 24.9, 23.9, 23.6. HRMS (ESI) for C13H18O4Na [M + Na]+ found 261.1097 calcd 261.1096.
(R)-6-Vinyl-5,6-dihydro-2H-pyran-2-one (8). Compound 20 (0.545 g, 2.28 mmol) was dissolved in 80% aq CF3COOH (1 mL) at 0 °C and the mixture was stirred at the same temperature for 2 h. The reaction mixture was then extracted with CH2Cl2 (3 × 5 mL). The collected organic layers were combined, washed with 10% NaHCO3 (3 × 5 mL), water, and brine, dried over Na2SO4, and concentrated in vacuo. The crude product was taken as such for the next step without further purification. To a solution of crude diol (0.3 g, 1.89 mmol) in dry toluene (5 mL) was added triphenylphosphine (1.989 g, 7.59 mmol) followed by imidazole (0.517 g, 7.59 mmol) and stirred vigorously. To the resulting solution was added iodine (0.722 g, 5.69 mmol) and the mixture was refluxed at 110 °C for 3 h. The reaction mixture, after bringing to room temperature, was decanted into excess sat. aq Na2S2O3 (5 mL) and sat. aq NaHCO3 (5 mL) in a separatory funnel. The residue in the reaction flask was extracted with EtOAc (3 × 5 mL). These extracts were combined with the material in the separatory funnel and shaken until the iodine was consumed. The organic phase was washed with H2O (1 × 5 mL), dried, and concentrated. The crude residue was chromatography (30% hexane/EtOAc), to obtain 8 (0.193 g, 68% (over 2 steps) as liquid. [α]25D: +80.6 (c 0.9, CHCl3). IR (neat) νmax: 1723, 1385, 1248, 1033, 817 cm−1. 1H NMR (500 MHz, CDCl3): δ 6.88 (ddd, J = 9.7, 5.4, 3.0 Hz, 1H), 6.04 (td, J = 9.7, 1.2 Hz, 1H), 5.94 (ddd, J = 17.0, 10.6, 5.7 Hz, 1H), 5.40 (dd, J = 17.2, 0.9 Hz, 1H), 5.29 (dd, J = 10.5, 0.9 Hz, 1H), 4.95–4.90 (m, 1H), 2.51–2.38 (m, 2H). 13C NMR (CDCl3, 75 MHz): δ 163.3, 144.0, 134.2, 120.9, 117.3, 76.1, 28.7. HRMS (ESI) for C7H8O2Na [M + Na]+ found 147.0422 calcd 147.0422.
(R)-((3-(Benzyloxy)hex-5-en-1-yl)oxy)(tert-butyl)diphenylsilane (14). To a stirring solution of alcohol 12 (2 g, 5.64 mmol), freshly prepared benzyl trichloroacetimidate (2.14 g, 8.47 mmol), and CH2Cl2 (10 mL) in a 50 mL round bottom flask, under an atmosphere of N2, was added (±)-camphor-10-sulfonic acid (131 mg, 0.56 mmol) in one portion. The reaction was allowed to proceed for 12 h at rt, after which time TLC analysis indicated essentially complete consumption of starting material. The reaction mixture was concentrated under reduced pressure, diluted with 20% hexane/EtOAc (30 mL), filtered over a pad of celite, and concentrated under reduced pressure to give a red slurry. Purification was accomplished by column chromatography (5% hexane/EtOAc), collecting 8 mL fractions. The product containing fractions were combined and concentrated under reduced pressure to give benzyl ether 14 (2.1 g, 84% yield) as colorless oil. [α]25D: +13.8 (c 1.5, CHCl3). IR (neat) νmax: 3069, 2956, 2932, 2891, 1639, 1427, 1109, 735, 701 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.72–7.69 (m, 4H), 7.44–7.33 (m, 11H), 5.88–5.79 (m, 1H), 5.10–5.04 (m, 2H), 4.78 (s, 2H), 3.86–3.78 (m, 1H), 3.77–3.70 (m, 2H), 2.36–2.31 (m, 2H), 1.80–1.75 (m, 2H), 1.10 (s, 9H). 13C NMR (CDCl3, 75 MHz): δ 138.4, 135.8, 135.1, 134.7, 134.5, 129.6, 129.5, 127.6, 127.5, 127.4, 127.3, 72.6, 70.2, 66.8, 41.5, 36.0, 26.5, 18.9. HRMS (ESI) for C29H36O2SiNa [M + Na]+ found 467.2376 calcd 467.2373.
(4S,6S)-6-(Benzyloxy)-8-((tert-butyldiphenylsilyl)oxy)oct-1-en-4-ol (15). To a solution of 14 (2.01 g, 4.52 mmol) in 1,4-dioxane/water (3:1; 12 mL), 2,6-lutidine (1.1 mL, 9.05 mmol), OsO4 (4.6 mL, 0.09 mmol) followed by NaIO4 (3.87 g, 18.1 mmol) were sequentially added at room temperature, and the mixture was stirred for 2 h. After completion of the reaction (monitored by TLC), 1,4-dioxane was removed under reduced pressure, and the residue was diluted with CH2Cl2 (20 mL). The organic layer was separated and the aqueous layer extracted with CH2Cl2 (2 × 10 mL). The combined organic layers were quickly washed with 1 N HCl (2 × 5 mL) to remove excess 2,6-lutidine followed by brine (2 × 5 mL), dried with anhydrous Na2SO4, and concentrated under reduced pressure to give the crude aldehyde. To a solution of crude aldehyde (1.8 g, 4.03 mmol) in CH2Cl2 (15 mL) at 0 °C were added MgBr2·OEt2 (2.08 g, 8.07 mmol), and allyltrimethylsilane (3.2 mL, 20.17 mmol). The resultant mixture was stirred at 0 °C overnight before being quenched with 1 N aq HCl solution. The resultant mixture was warmed to room temperature and extracted with EtOAc. The organic layer was washed successively with sat. aq NaHCO3 solution and brine. The organic layer was dried (Na2SO4), filtered, and concentrated under reduced pressure. Purification of the residue by column chromatography silica gel (20% hexane/EtOAc) to give homoallylic alcohol 15 (1.693 g, 76.5% (over 2 steps) as a colorless oil. [α]25D: +25.6 (c 1.3, CHCl3). IR (neat) νmax: 3432, 3069, 2932, 2857, 1639, 1427, 1108, 702 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.68–7.63 (m, 4H), 7.45–7.25 (m, 1H), 5.85–5.75 (m, 1H), 5.12–5.06 (m, 2H), 4.5 (s, 2H), 4.00–3.89 (m, 2H), 3.83–3.69 (m, 2H), 2.23–2.16 (m, 2H), 2.00–1.92 (m, 1H), 1.80–1.70 (m, 1H), 1.63–1.57 (m, 2H), 1.05 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 138.1, 135.5, 134.8, 134.7, 133.6, 129.6, 128.3, 127.8, 127.6, 117.4, 74.2, 71.4, 67.7, 60.4, 42.1, 39.5, 36.5, 26.8, 19.1. HRMS (ESI) for C31H40O3 SiNa [M + Na]+ found 511.2638 calcd 511.2623.
(3S,5S)-1-((tert-Butyldiphenylsilyl)oxy)oct-7-ene-3,5-diol (16). To a stirred solution of naphthalene (2.6 g, 20.28 mmol) in THF (10 mL) were added lithium granules (165 mg, 23.66 mmol) at room temperature, and the solution was allowed to stir at room temperature for 30 min to generate Li naphthalenide. To the resulting dark green solution was added benzyl ether 15 (1.65 g, 3.38 mmol) at −10 °C, and the mixture was allowed to stir at the same temperature for 30 min, quenched with aqueous NH4Cl, extracted into EtOAc (3 × 50 mL), dried over Na2SO4, concentrated, and purified on silica gel (40% hexane/EtOAc) to give diol 16 (1.143 g, 85%) as a colorless liquid. [α]25D: −14.9 (c 1.2, CHCl3). IR (neat) νmax: 3416, 2931, 2857, 1428, 1109, 1083, 740, 704 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.69–7.65 (m, 4H), 7.47–7.37 (m, 6H), 5.89–5.79 (m, 1H), 5.16–5.09 (m, 2H), 4.30–4.22 (m, 1H), 4.05–3.98 (m, 1H), 3.90–3.85 (m, 2H), 3.78 (br.s, 1H), 2.30–2.25 (m, 2H), 1.92–1.78 (m, 1H), 1.70–1.56 (m, 3H), 1.05 (s, 9H). 13C NMR (CDCl3, 75 MHz): δ 135.4, 134.8, 132.8, 132.6, 129.8, 127.7, 117.6, 69.5, 67.9, 63.6, 42.2, 42, 38.1, 26.7, 18.9. HRMS (ESI) for C24H34O3SiNa [M + Na]+ found 421.2169 calcd 421.2158.
(2-((4S,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)ethoxy)(tert-butyl)diphenylsilane (17). 2,2-Dimethoxypropane (0.7 mL, 5.52 mmol) and catalytic PPTS (83 mg, 0.33 mmol) were added successively to a solution of diol 16 (1.1 g, 2.76 mmol) in a CH2Cl2 (8 mL). The solution was stirred for 10 h at room temperature and then quenched with solid NaHCO3. The crude compound was concentrated in vacuo and purified by column chromatography (15% hexane/EtOAc) to afford the acetonide product 17 (1.137 g, 94%) as a colorless liquid. [α]25D: +27.8 (c 0.8, CHCl3). IR (neat) νmax: 2934, 2858, 1379, 1223, 1110, 739, 704 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.69–7.63 (m, 4H), 7.44–7.35 (m, 6H), 5.84–5.75 (m, 1H), 5.13–5.02 (m, 2H), 4.13–4.05 (m, 1H), 3.89–3.75 (m, 2H), 3.72–3.66 (m, 1H), 2.36–2.88 (m, 1H), 2.22–2.15 (m, 1H), 1.77–1.54 (m, 4H), 1.35 (s, 3H), 1.33 (s, 3H), 1.04 (s, 9H). 13C NMR (CDCl3, 75 MHz): δ 135.4, 134.4, 133.8, 129.5, 127.5, 116.7, 100.1, 66.1, 63.3, 60, 40.1, 38.8, 38, 26.8, 24.8, 19.1. HRMS (ESI) for C27H38O3SiNa [M + Na]+ found 461.2482 calcd 461.2471.
2-((4S,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)ethanol (11). A 1 M solution of TBAF in THF (4.56 mL, 4.56 mmol) was added to a solution of compound 17 (1 g, 2.28 mmol) in dry THF (5 mL) at 0 °C. The mixture was stirred at room temperature for 3 h. After completion of the reaction, the mixture was diluted with EtOAc (15 mL). The combined organic layers were washed with brine, and the mixture was extracted with EtOAc (3 × 10 mL), dried over Na2SO4. The solvent was removed under reduced pressure, and the mixture was purified by column chromatography (20% hexane/EtOAc) to afford 11 (410 mg, 90%) as a colorless liquid. [α]25D: +40.6 (c 1.6, CHCl3). IR (neat) νmax: 3422, 2987, 2939, 1643, 1308, 1224, 1168, 1054, 761 cm−1. 1H NMR (500 MHz, CDCl3): δ 5.83–5.73 (m, 1H), 5.12–5.03 (m, 2H), 4.08–4.01 (m, 1H), 3.91–3.84 (m, 1H), 3.78–3.72 (m, 2H), 2.55 (br.s, 1H), 2.34–2.77 (m, 1H), 2.23–2.16 (m, 1H), 1.77–1.70 (m, 2H), 1.69–1.61 (m, 2H), 1.37 (s, 3H), 1.35 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 134.2, 117, 100.4, 66.5, 66.1, 40, 37.6 (2C), 24.8, 24.7. HRMS (ESI) for C11H20O3Na [M + Na]+ found 223.1312 calcd 223.1310.
(R)-1-((4S,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)-4-phenylbut-3-yn-2-ol (9). To the solution of alcohol 11 (0.3 g, 1.50 mmol) in anhydrous CH3CN (5 mL) IBX (0.63 g, 2.25 mmol) was added and the resulting suspension was vigorously stirred at reflux condition for 1 h, and the reaction was allowed to cool to rt, filtered through celite. The residue was washed with EtOAc (3 × 5 mL) and the combined filtrate was concentrated to give the crude aldehyde, which was directly used for the next reaction without further purification. In 25 mL flask, 1 mL toluene solution of phenylacetylene (0.56 mL, 5.09 mmol) and diethylzinc (5.1 mL, 5.09 mmol) was refluxed for 1 hour under nitrogen. After the solution was cooled to room temperature, (S)-BINOL (360 mg, 1.27 mmol), diethyl ether (8 mL) and Ti(OiPr)4 (0.37 mL, 1.27 mmol) were added sequentially. The solution was stirred for another 1 h, and aldehyde (252 mg, 1.27 mmol) was added. After additional 4 h, the reaction was quenched with saturated ammonium chloride. The resulting mixture was extracted with CH2Cl2 and concentrated under vacuum. Purification of the residue by passing through a short silica gel column (20% hexane/EtOAc) afforded the pure propargylic alcohol product 9 (335 mg, 74.8%, over 2 steps) as a colorless liquid. [α]25D: +9.1 (c 0.6, CHCl3). IR (neat) νmax: 3412, 3078, 2986, 2926, 1638, 1490, 1381, 1223, 1025, 756 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.44–7.41 (m, 2H), 7.33–7.28 (m, 3H), 5.84–5.75 (m, 1H), 5.13–5.04 (m, 2H), 4.48–4.79 (m, 1H), 4.18–4.11 (m, 1H), 3.90 (qt, J = 14.1, 7.0 Hz, 1H), 2.98 (d, J = 2.7 Hz, 1H), 2.35–2.28 (m, 1H), 2.24–2.17 (m, 1H), 2.10–2.02 (m, 1H), 1.97–1.91 (m, 1H), 1.70 (t, 2H), 1.40 (s, 3H), 1.37 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 134.2, 131.6, 128.3, 128.2, 117, 100.6 (2C), 89.3, 66.1, 66, 43.2, 40, 37.8, 24.9, 24.6. HRMS (ESI) for C19H24O3Na [M + Na]+ found 323.1623 calcd 323.1621.
(R)-(R,E)-1-((4R,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)-4-phenylbut-3-en-2-yl-2-methoxy-2-phenylacetate. To a solution of propargylic alcohol 9 (12 mg, 0.066 mmol) in dry CH2Cl2 (5 mL), DMAP catalytic, (R)-O-methylmandelic acid (9 mg, 0.079 mmol) and DCC (12 mg, 0.099 mmol) were added and the mixture was stirred at room temperature for 60 minutes under argon. The reaction was diluted with DCM and filtered through celite. The filtrate was concentrated under reduced pressure and purified by column chromatography (5–10% hexane/ethyl acetate) to afford O-methylmandelate as a colorless oil (15 mg, 85%). 1H NMR (500 MHz, CDCl3): δ 7.49–7.43 (m, 2H), 7.38–7.26 (m, 8H), 5.82–5.74 (m, 1H), 5.11–5.03 (m, 2H), 4.81 (s, 1H), 4.13–4.06 (m, 1H), 3.89–3.82 (m, 1H), 3.45 (s, 3H), 3.44–3.40 (m, 1H), 2.33–2.25 (m, 1H), 2.21–2.14 (m, 1H), 2.07–1.92 (m, 2H), 1.66–1.58 (m, 2H), 1.35 (s, 3H), 1.33 (s, 3H).
(S)-(R,E)-1-((4R,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)-4-phenylbut-3-en-2-yl-2-methoxy-2-phenylacetate. To a solution of propargylic alcohol 9 (10 mg, 0.066 mmol) in dry CH2Cl2 (5 mL), DMAP catalytic, (S)-O-methylmandelic acid (6 mg, 0.079 mmol) and DCC (10 mg, 0.099 mmol) were added and the mixture was stirred at room temperature for 60 minutes under argon. The reaction was diluted with DCM and filtered through celite. The filtrate was concentrated under reduced pressure and purified by column chromatography (5–10% hexane/ethyl acetate) to afford O-methylmandelate as a colorless oil (13 mg, 87%). 1H NMR (500 MHz, CDCl3): δ 7.50–7.27 (m, 10H), 5.79–5.70 (m, 1H), 5.10–5.01 (m, 2H), 4.81 (s, 1H), 4.17–4.07 (m, 1H), 4.02–3.95 (m, 1H), 3.82–3.76 (m, 1H), 3.43 (s, 3H), 2.34–2.32 (m, 1H), 2.17–2.10 (m, 1H), 1.92–1.8 (m, 1H), 1.65–1.50 (m, 3H), 1.30 (s, 3H), 1.29 (s, 3H).
(R,E)-1-((4S,6S)-6-Allyl-2,2-dimethyl-1,3-dioxan-4-yl)-4-phenylbut-3-en-2-ol (18). To a cold (0 °C) solution of propargylic alcohol 9 (0.2 g, 0.66 mmol) in THF (5 mL) was added Red-Al (0.32 mL, 70 wt% in toluene, 0.99 mmol) dropwise. After 3 h at 0 °C, the reaction mixture was quenched carefully by dropwise addition of saturated aqueous sodium sulphate (Caution: vigorous evolution of H2 may result). EtOAc was added and the mixture was allowed to warm to rt. The organic layer was washed with brine and the combined aqueous layer was extracted several times with EtOAc. The combined organic extracts were dried over Na2SO4, filtered and concentrated under reduced pressure. The residue was chromatographed on silica gel (30% hexane/EtOAc) to provide (187 mg, 93% yield) of allylic alcohol 18 as a colorless oil. [α]25D: +24.5 (c 0.9, CHCl3). IR (neat) νmax: 3437, 3026, 2986, 2937, 1641, 1449, 1380, 1223, 1166, 1123, 968, 749 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.42–7.18 (m, 5H), 6.61 (d, J = 15.8 Hz, 1H), 6.18 (dd, J = 15.8, 6.4 Hz, 1H), 5.87–5.71 (m, 1H), 5.14–5.02 (m, 2H), 4.53–4.44 (m, 1H), 4.16–4.03 (m, 1H), 3.95–3.84 (m, 1H), 3.43 (br.s, 1H), 2.37–2.14 (m, 2H), 1.83–1.57 (m, 4H), 1.41 (s, 3H), 1.38 (s, 3H). 13C NMR (CDCl3, 75 MHz): δ 136.7, 134.1, 131.6, 129.8, 128.4, 127.4, 126.3, 117, 100.6, 72.2, 66.9, 66, 42.7, 39.9, 38, 24.9, 24.6. HRMS (ESI) for C19H26O3Na [M + Na]+ found 325.1779 calcd 235.1770.
(3R,5R,7S,E)-1-Phenyldeca-1,9-diene-3,5,7-triol (7). To a solution that was cooled in an ice bath of compound 18 (0.1 g, 0.33 mmol) in CH3CN (5 mL) was added CuCl2·2H2O (112 mg, 0.66 mmol) as a solid. The reaction mixture was stirred at 0 °C for 1 h. The reaction mixture was quenched with solid NaHCO3 (2 g), and the resulting mixture was filtered. The filtrate was concentrated under reduced pressure, and the crude product was purified by column chromatography (60% hexane/EtOAc) to afford triol 7 (78 mg, 90%) as a colorless oil. [α]25D: +42.7 (c 0.5, CHCl3). IR (neat) νmax: 3369, 3080, 3027, 2939, 1640, 1433, 1070, 967, 749 cm−1. 1H NMR (300 MHz, CDCl3): δ 7.43–7.16 (m, 5H), 6.60 (d, J = 15.8 Hz, 1H), 6.22 (dd, J = 15.8, 6.7 Hz, 1H), 5.89–5.73 (m, 1H), 5.20–5.08 (m, 2H), 4.63–4.53 (m, 1H), 4.35–4.23 (m, 1H), 4.02 (qt, J = 12.8, 6.7 Hz, 1H), 2.32–2.22 (m, 2H), 1.90–1.59 (m, 4H). 13C NMR (CDCl3, 75 MHz): δ 136.5, 134.5, 131.6, 129.8, 128.5, 127.6, 126.4, 117.9, 73, 69.4, 67.7, 43.1, 42.4, 42. HRMS (ESI) for C16H22O3Na [M + Na]+ found 285.1461 calcd 285.1461.
(R)-6-((1E,4S,6R,8R,9E)-4,6,8-Trihydroxy-10-phenyldeca-1,9-dien-1-yl)-5,6-dihydro-2H-pyran-2-one (6) (cryptomoscatone F1). A solution of Grubbs II catalyst (3 mg, 0.003 mmol) in CH2Cl2 (1 mL) was added dropwise to a solution of the triol 7 (16 mg, 0.061 mmol) and vinyl lactone 8 (11 mg, 0.091 mmol) in CH2Cl2 (1 mL) at rt, and the mixture was refluxed for 1 h. The solvent was removed under reduced pressure and the crude product was purified by column chromatography (80% hexane/EtOAc) to give viscous liquid (19 mg, 89%). [α]25D: +35.0 (c 1.0, CHCl3). IR (neat) νmax: 3405, 2925, 2854, 1713, 1384, 1250, 1054, 753 cm−1. 1H NMR (500 MHz, CDCl3): δ 7.41–7.22 (m, 5H), 6.91–6.85 (m, 1H), 6.60 (d, J = 15.8 Hz, 1H), 6.23 (dd, J = 15.8, 6.7 Hz, 1H), 6.03 (dt, J = 9.9, 1.6 Hz, 1H), 5.92–5.84 (m, 1H), 5.69 (dd, J = 15.5, 6.5 Hz, 1H), 4.93–4.86 (m, 1H), 4.62–4.56 (m, 1H), 4.33–4.24 (m, 1H), 4.08–4.01 (m, 1H), 2.47–2.39 (m, 2H), 2.35–2.22 (m, 2H), 1.92–1.82 (m, 1H), 1.75–1.60 (m, 3H).13C NMR (CDCl3, 125 MHz): δ 164.2, 144.9, 136.5, 131.6, 131.4, 130, 129.5, 128.5, 127.6, 126.4, 121.3, 77.9, 73.3, 69.6, 67.9, 42.9, 42.4, 40.2, 29.6. HRMS: calcd for C21H26O5Na [M + NH4]+: 381.1678: found: 381.1674.
Acknowledgements
AR thank UGC, New Delhi for the award of fellowship. All the authors thank CSIR, New Delhi for financial support as part of XII Five Year plan programme under title ORIGIN (CSC-0108).
References
- C. J. Nehme, W. L. Bastos, A. J. de Araújo and A. J. Cavalheiro, Phytochem. Anal., 2005, 16, 93–97 CrossRef CAS PubMed.
- A. J. Cavalheiro and M. Yoshida, Phytochemistry, 2000, 53, 811–819 CrossRef CAS.
-
(a) A. de Fatima, L. V. Modolo, L. S. Conegero, R. A. Pilli, C. V. Ferreira, L. K. Kohn and J. E. de Carvalho, Curr. Med. Chem., 2006, 13, 3371–3384 CrossRef CAS;
(b) C. M. Sturgeon, B. Cinel, A. R. Díaz-Marrero, L. M. McHardy, M. Ngo, R. J. Andersen and M. Roberge, Cancer Chemother. Pharmacol., 2008, 61, 407–413 CrossRef CAS PubMed.
- M. P. Giocondo, C. L. Bassi, M. Telascrea, A. J. Cavalheiro, V. S. Bolzani, D. H. S. Silva, D. Agustoni, E. R. Mello and C. P. Soares, Rev. Cienc. Farm. Basica Apl., 2009, 30, 315–322 CAS.
- G. Schmeda-Hirschmann, L. Astudillo, J. Bastida, C. Codina, A. R. De Arias, M. E. Ferreira, A. Inchaustti and G. Yaluff, J. Pharm. Pharmacol., 2001, 53, 563–567 CrossRef CAS.
-
(a) A. Raju and G. Sabitha, Tetrahedron Lett., 2014, 55, 5756–5758 CrossRef CAS PubMed;
(b) L. F. Toneto Novaes, R. L. Drekener, C. M. Avila and R. A. Pilli, Tetrahedron, 2014, 70, 6467–6473 CrossRef CAS PubMed;
(c) G. Chinna Reddy, P. Balasubramanyam, N. Salvanna, T. Sreenivasulu Reddy and B. Das, Bioorg. Med. Chem. Lett., 2012, 22, 2415–2417 CrossRef CAS PubMed;
(d) J. S. Yadav, D. C. Bhunia, B. Ganganna and V. K. Singh, RSC Adv., 2013, 3, 5254–5260 RSC.
- G. Sabitha, S. Siva Sankara Reddy, D. Vasudeva Reddy, V. Bhaskar and J. S. Yadav, Synthesis, 2010, 20, 3453–3460 CrossRef PubMed.
- A. Chattopadhyay and V. R. Mamdapur, J. Org. Chem., 1995, 60, 585–587 CrossRef CAS.
- G. E. Keck and A. P. Truong, Org. Lett., 2005, 7, 2153–2156 CrossRef CAS PubMed.
- W. Yu, Y. Mei, Y. Kang, Z. Hua and Z. Jin, Org. Lett., 2004, 6, 3217–3219 CrossRef CAS PubMed.
- H. Fuwa and M. Sasaki, Org. Lett., 2010, 12, 584–587 Search PubMed.
- S. D. Rychnovsky, B. Rogers and G. Yang, J. Org. Chem., 1993, 58, 3511–3515 CrossRef CAS.
- G. Gao, D. Moore, R.-G. Xie and L. Pu, Org. Lett., 2002, 4, 4143–4146 CrossRef CAS PubMed.
- The diastereomeric ratio of the product was determined using a Shimadzu high-performance liquid chromatography (HPLC) system equipped with a chiral HPLC column (XDB C18, 150 × 4.6, 5U) and a UV detector at an absorbance of 225 nm. ZORBAX SBC 18 250 × 4.6 mm, 5 m (column) and a solvent system of acetonitrile and water (3:1) at a flow rate of 1.0 mL per min were used. tR: 21.17 and 24.34 min.
- J. De Brabander and M. Vandewalle, Synthesis, 1994, 8, 856–865 Search PubMed.
- The diastereomeric ratio of the product was determined using a Shimadzu high-performance liquid-chromatography (HPLC) system equipped with a chiral HPLC column (Chiralcel OD) and a UV detector at an absorbance of 254 nm. A solvent system of acetonitrile and water (4:6) at a flow rate of 1.0 mL min−1 was used. tR: 5.74 and 5.89 min.
- V. K. Yadav and D. Agrawal, Chem. Commun., 2007, 5232–5234 RSC.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953–956 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectral data of all compounds and copies of 1H and 13C NMR spectra of all compounds. See DOI: 10.1039/c5ra03693c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 







