
Enhancement of photoluminescence emission and anomalous photoconductivity properties of Fe3O4@SiO2 core–shell microspheres
S. K. Jana†
*a,
S. Majumder†a,
B. Satpatia,
S. K. Mishrab,
R. K. Srivastavac and
S. Banerjeea
aSurface Physics and Material Science Division, Saha Institute of Nuclear Physics, 1/AF Bidhannagar, Saltlake, Kolkata-700064, India. E-mail: skj_ec@yahoo.co.in
bDepartment of Physics, University of Lucknow, Lucknow-226007, India
cDepartment of Electronics and Communication, University of Allahabad, Allahabad-211002, India
First published on 17th March 2015
Abstract
In this manuscript we report the successful synthesis of both pristine Fe3O4 and the Fe3O4@SiO2 core@shell structure. From SEM images we observe that each Fe3O4 microsphere is composed of a large number of smaller nanoballs. We have extensively studied the photoluminescence and photoconductivity properties of both pristine and SiO2 coated Fe3O4 particles for the first time. An enhancement in photoluminescence emission is observed in the Fe3O4@SiO2 core@shell samples, whereas a reduced and negative photoconductivity is observed in the same sample. SiO2 coating reduces the concentrations of non-radiative trap levels at the interfaces of the core and shell, thereby resulting in the enhancement of photoluminescence intensity in the core–shell particles. An exponential rise and decay in photocurrent is observed upon UV irradiation in the ON and OFF state, respectively, for Fe3O4, whereas for Fe3O4@SiO2, we observe a transient rise in the photocurrent and this photocurrent is not stable. We have explained this unusual behavior of photocurrent.
(1) Introduction
Recently core–shell structured nanoparticles have received considerable attention due to their modified optical, electronic and magnetic properties compared to those of single-component nanomaterials.1 Among all the currently studied nanomaterials, Fe3O4 is one of the most popular and has been found to have large applications in information storage, magnetic refrigeration, magneto optical solid devices, cell separation and magnetic resonance imaging enhancement.2–4 The structural and magnetic properties of SiO2 coated Fe3O4 have been discussed elaborately in some earlier reports,5–7 where the SiO2 coating not only provides a chemically inert surface for the magnetic nanoparticles but also allows the nanoparticle to conjugate on its surface with various functional groups. Because the surface defects of nanostructured materials are known to have a great influence on their optical properties,8–10 studies of the photo response of both the Fe3O4 and Fe3O4@SiO2 core–shell system have drawn considerable attention in the photonic research field. The photocatalytic properties of Fe3O4 and Fe3O4@SiO2 core–shell particles at a high energy of incident spectrum5–7 have been reported but investigations of PL and PC on such samples have not been reported to date. In this manuscript we report the effect of the SiO2 shell on the PL and PC of these materials.Several groups have investigated the photoconductivity of silica based nanocomposite systems.11–14 Enhanced photoconductivity has also been observed for SiO2 based ZnO nanocomposites,11–13 and a reduced photoconductivity was reported for ZnO quantum dots embedded in a SiO2 matrix.14 The PC study depends exclusively on the surface properties of the nanomaterials. Various explanations have been proposed to explain the PC of different oxide materials.15–17 Broadly, two different mechanisms have been proposed for the origin of PC: the first is a fast band to band transition with characteristic time in the nanosecond range and the second is the adsorption/desorption of oxygen molecules at the interfaces of the nanomaterials (where the holes generated upon illumination help to discharge oxygen species from the surfaces by an indirect electron hole recombination mechanism14).
In this work, we have studied for the first time the unusual optical properties of Fe3O4 based core–shell particles. Both pristine Fe3O4 and Fe3O4@SiO2 core–shell structures have been successfully synthesized. We have investigated how the surface morphology of Fe3O4 affects its PL and PC properties. In addition, we have discussed in detail the influence of the SiO2 coating i.e., the surface properties on the PL and PC of the Fe3O4@SiO2 core–shell particles.
(2) Experimental
The core–shell microsphere sample was synthesized using a two-step method: (i) the synthesis of Fe3O4 microspheres (core formation) and (ii) coating the Fe3O4 microsphere with SiO2 (shell formation). 2.7 g of iron chloride(III) hexahydrate, 2.0 g of polyethylene glycol (PEG, Mw = 4000) and 7.2 g of sodium acetate trihydrate were added into 80 mL ethylene glycol under constant magnetic stirring. Then, the solution was transferred into a Teflon-lined stainless steel autoclave with a capacity of 100 mL and heated at 180 °C for 19 h. The product was collected, washed with de-ionized (DI) water and absolute ethanol, and finally dried at 65 °C for 3 h. For the preparation of SiO2 coated particles, 0.4 g of the as-prepared Fe3O4 was dispersed into a mixture of 40 mL ethanol and 8 mL DI water in a glass beaker under constant stirring. Then, 2 mL of ammonia solution (25 wt%) and 1.6 mL of tetraethyl orthosilicate (TEOS) were consecutively added and the mixture was kept under constant stirring for 3 h. Finally, the resultant products were collected, washed and dried at 65 °C for 3–6 h.Structural analysis of both pristine Fe3O4 and Fe3O4@SiO2 core–shell material was carried out by X-ray diffraction (Bruker AXS D8) using Cu Kα radiation. The surface morphology of both pristine Fe3O4 and the Fe3O4@SiO2 core–shell was obtained using scanning electron microscopy (SEM) measurements, and detailed structural information was obtained using transmission electron microscopy (TEM: FEI, TF30, ST microscope operating at 300 kV). The TEM is equipped with a scanning unit and a high angle annular dark-field (HAADF) detector obtained from Fischione (model: 3000). The samples were mounted on a carbon coated Cu grid and used for TEM measurements.
Room temperature photoluminescence (PL) measurements were obtained for both samples having same concentration of aqueous solution using a JASCO spectrofluorometer (FP 6700) at an excitation of 320 nm, keeping a fixed slit width of 2.5 nm in both cases.
The photo and dark conductivities of both pristine and SiO2 coated Fe3O4 NPs were measured using a thick film of samples in-between interdigitated Cu electrodes. The two electrodes topped with a thick sample were pressed with a transparent glass plate. This glass plate had a slit for providing an illumination area of 0.25 cm2. In this cell type device, the direction of illumination was normal to the field across the electrodes. The cell was mounted in a dark chamber equipped with a slit from where light was allowed to fall over the cell. The photoresponse was measured with a 300 W mercury lamp under UV illumination at 365 nm of fixed intensity as an excitation source. A stabilized dc field (5 V cm−1 to 50 V cm−1) was applied across the cell, and both dark current and photocurrent were measured by a nanoammeter (NM122, Scientific Equipment) in series with a RISH 15S multimeter connected with a RISH Multi SI 232 adapter. Before measuring the photoconductivity of the sample, the cell was first kept in dark till it attained equilibrium.
(3) Results and discussion
The structure and morphology of pristine Fe3O4 and Fe3O4@SiO2 were analyzed using both SEM and TEM images, as shown in Fig. 1 and 2, respectively. From Fig. 1a, it is observed that the Fe3O4 microsphere is formed from a large number of Fe3O4 nanoballs, and Fig. 1b reveals the core–shell formation of Fe3O4@SiO2, where Fe3O4 microsphere appears as the core and a thin layer of SiO2 as the shell. Moreover, it is confirmed that a large number of PEG capped Fe3O4 nanoparticles agglomerate to form a Fe3O4 microsphere, as shown in Fig. 1c. | ||
| Fig. 1 SEM morphology of (a) Fe3O4, (b) Fe3O4@SiO2 and (c) the formation of Fe3O4 microspheres from a large number of Fe3O4 nanoballs. | ||
 | ||
| Fig. 2 (a)–(b) TEM images and (c) STEM-HAADF image of the Fe3O4 microspheres. (d) TEM image and (e) STEM-HAADF image of Fe3O4@SiO2 microspheres. (f) Selected area (‘2’) EDX analysis and (g) line scan EDX analysis of Fe3O4@SiO2. (h) Elemental color mapping of the Fe3O4@SiO2 core–shell samples. | ||
Fig. 2a shows the TEM image of the pristine Fe3O4 microspheres, which is consistent with the SEM image of the pristine sample. In addition, the high resolution TEM image (shown in Fig. 2b) confirms that the Fe3O4 microspheres consist of smaller Fe3O4 nanoparticles. We observe that the arrangement of Fe3O4 nanoparticles inside a microsphere creates some voids spaces, which can be seen in the inset of Fig. 2b. The high angle annular dark-field scanning transmission electron microscopy (STEM-HAADF) image (shown in Fig. 2c) of Fe3O4 is consistent with the SEM image of the same sample shown in Fig. 1a. Both the TEM image and STEM-HAADF image shown in Fig. 2d and e demonstrate the successful synthesis process of the core–shell particles. The energy dispersive X-ray (EDX) spectrum, shown in Fig. 2f, from area 2 in the STEM-HAADF image (Fig. 2e) and subsequent line scan along line 1 and elemental map from area 2 (Fig. 2g and h, respectively) confirmed that the core material is Fe3O4 and SiO2 is formed as shell.
The formation mechanism of bare and SiO2 coated Fe3O4 (core–shell) microspheres is shown schematically in Fig. 3. TEM images are also shown individually below each step in the formation mechanism in the schematic. In the first step, we have synthesized PEG capped Fe3O4 NPs of sizes ∼18 nm (first TEM image of the schematic) by a co-precipitation method. In addition, the HRTEM image of the individual Fe3O4 NPs shows lattice fringes, which prove that the NPs are crystalline in nature. In the second step, there are a large number of Fe3O4 NPs that agglomerate to form the Fe3O4 microspheres, as shown in the second TEM image in the schematic. In the last step, a thin layer of SiO2 cell has been formed on Fe3O4 microspheres by the hydrolysis of TEOS followed by cross linking with Fe3O4, as shown in the schematic diagram, and we obtained the Fe3O4@SiO2 core–shell particles, which are shown in the TEM image. The mechanism behind the formation of the Fe3O4 microspheres is that the magnetite nanocrystals nucleate first in a supersaturated solution, which is the solvent-mediated hydrolysis of Fe3+. Afterwards, the newly formed nanocrystals aggregate into round spheres, which is driven by the minimization of the total surface energy.18
 | ||
| Fig. 3 Schematic representation of the formation of both Fe3O4 and Fe3O4@SiO2. | ||
The crystallographic phases of both Fe3O4 and the Fe3O4@SiO2 sample were identified by X-ray diffraction, as shown in Fig. 4. The diffraction peaks at 30.2°, 35.6°, 39.5°, 43.1°, 53.7°, 57.1° and 62.7° correspond to the [220], [311], [222], [400], [422], [511], and [440] planes of Fe3O4, which are consistent with the values mentioned in earlier reports.19,20 The positions of all the diffraction peaks of Fe3O4 powder are well-matched with the standard JCPDS 19-629. The diffraction pattern indicates that the Fe3O4 particles are in single phase and belong to a cubic system. For Fe3O4@SiO2, the small hump from 20.5° to 25° is due to the amorphous SiO2 shell, which is also consistent with earlier reports.20 Hence, it can be concluded that the SiO2 coating on Fe3O4 has not changed the crystal structure of the core Fe3O4 particles. The successful conjugation of SiO2 onto the surface of Fe3O4 was confirmed by Fourier transform infrared (FTIR) spectroscopy. The FTIR spectra of (a) Fe3O4 and (b) Fe3O4@SiO2 are shown in Fig. 5. In Fig. 5a, the band at 567 cm−1 is related to the Fe–O bending vibrations, whereas the band at 574 cm−1 in Fig. 5b is an indication of the presence of Si–O–Fe stretching vibrations.21
 | ||
| Fig. 4 XRD patterns of (a) Fe3O4 and (b) Fe3O4@SiO2. | ||
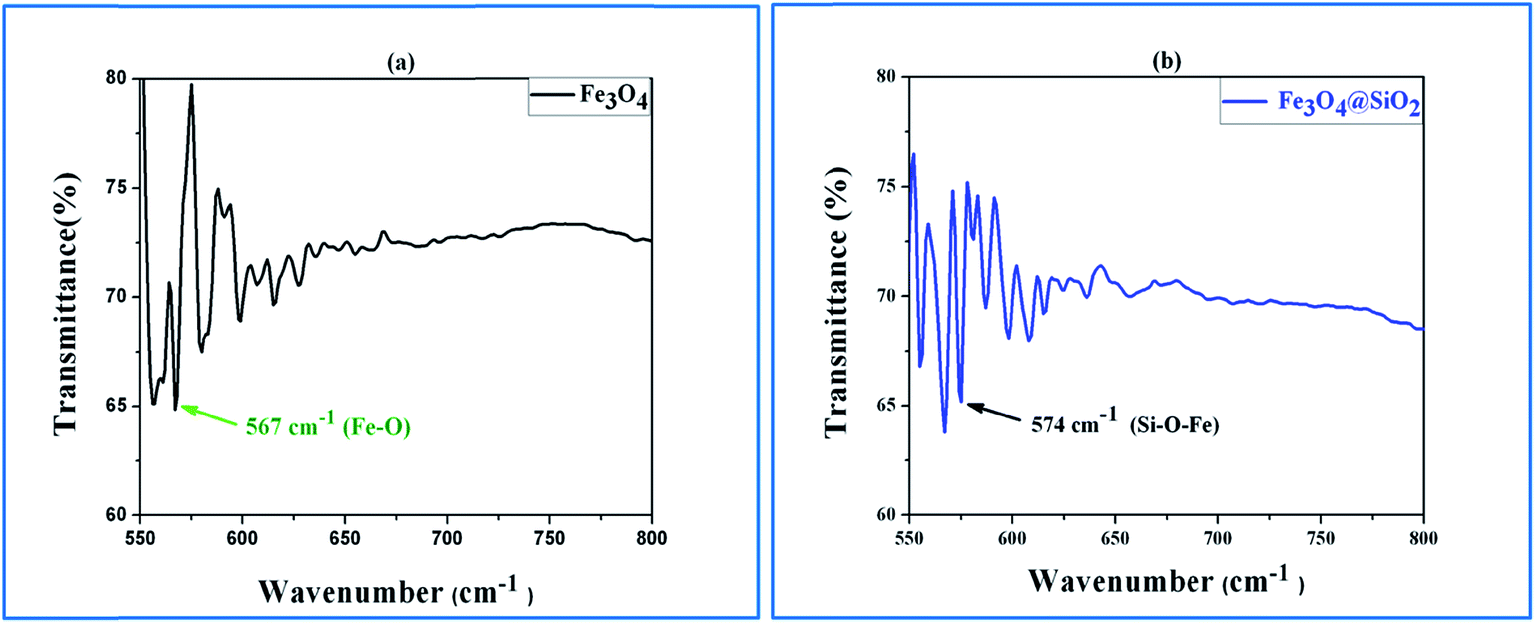 | ||
| Fig. 5 FTIR spectra of (a) Fe3O4 and (b) Fe3O4@SiO2. | ||
Photoluminescence excitation (PLE) measurements were performed keeping the emission wavelength fixed at 450 nm. The PLE curves for both the samples are shown in Fig. 6a. From this figure, it is observed that both the samples show a pronounced absorption peak between 305–400 nm, which is consistent with an earlier report22 on the absorption properties of the same samples. The peak position (∼327 nm) of the PLE spectra was attributed to the band edge excitation of both the samples. In addition, there is no peak shift in the PLE spectra for the core–shell structure. Therefore, we can say that there is no change in the band gap of the core–shell particle. However, the PLE spectra of the Fe3O4@SiO2 core–shell particles have a considerably sharper absorption edge (after 327 nm) when compared to pristine Fe3O4, which gives some indication of the modified surface properties of pristine Fe3O4 after SiO2 coating.
 | ||
| Fig. 6 (a) PLE measurements for both Fe3O4 (black line) and Fe3O4@SiO2 (blue line) samples. (b) Room temperature PL measurements for both Fe3O4 (black line) and Fe3O4@SiO2 (blue line) samples at an excitation wavelength of 320 nm. | ||
The PL emission peaks for both pristine Fe3O4 and Fe3O4@SiO2 samples at an excitation wavelength of 320 nm are shown in Fig. 6b and it can be seen that the emission wavelength at 414 nm is the same for both samples. The broad emission peak for Fe3O4 is observed because of oxygen vacancies, which play a key role in the origin of violet emission and is similar to the green PL observed for ZnO.8–10 The Fe3O4 nanoparticles have a very large surface area and thereby have a large number of dangling bonds associated with the oxygen vacancies, which constitutes their surface defects. These defects form donor states below the conduction band. The PL emission arises from the recombination of electrons in the donor states with photoexcited holes in the valence band. Furthermore, the Fe3O4 spheres show broadening in their PL emission with reduced intensity, which is a result of the scattering of incident and emitted photons from the highly roughened surface of Fe3O4. The SEM and TEM images show that the Fe3O4 spheres have surface voids and surface roughness, which are mostly caused by the agglomeration of small Fe3O4 particles. Upon SiO2 coating, the density of the surface dangling bonds was reduced due to the formation of Fe–O–Si in core–shell structure, as shown in the FTIR spectrum (Fig. 5). Hence, the SiO2 coating reduces the number of trap states and surface states. Thus, the probability of blue-green emission through radiative recombination is increased due to the reduction in the concentration of non-radiative trap levels.11,23 Thus, we observe that the coating of SiO2 on Fe3O4 enhances the PL property of Fe3O4. Moreover, the outer SiO2 layer decreases the surface voids as well as the roughness. Therefore, enhanced and coherent emission from the core–shell particle was obtained due to reduced light scattering by the SiO2 coating.
Fig. 7a and b show the I–V characteristics of the samples. The variation of dark current (Idc) and photocurrent (Ipc) with applied cell voltage on a log–log scale in the dark and under UV light illumination (λ = 365 nm) is shown for both pristine and core–shell Fe3O4 microspheres. These two graphs clearly represent the linear variation with different slopes and can be expressed by the power law i.e. I ∝ Vr, where I is either Ipc or Idc, V is the applied DC voltage and the exponent ‘r’ is the slope of straight line segment of the log–log plot. For the pristine Fe3O4 sample, the variation of Idc and Ipc with applied voltage is found to be non-ohmic super linear24–26 in nature for the entire voltage regime i.e. the power index is 1 < r < 2. The non-ohmic super-linear variation (1 < r < 2) in the dark current and photocurrent suggests that the charge carriers are being injected into the bulk of the materials produced from one of the electrodes.24 This photoresponse confirms the features of the sample itself, but not the features of the sample-contact region. The value of ‘r’ is different for the two samples and the highest value of ‘r’ is revealed by Fe3O4@SiO2 and may be attributed to the trapping of some of the photoexcited electrons in the SiO2 shell. From the I–V curves of both the pristine and core–shell samples, it is evident that the photocurrent for both the samples increases with voltage. It is clearly observed from Fig. 8 that the dark current in the Fe3O4@SiO2 sample is around ten times lower when compared to that in the bare Fe3O4 sample. A similar trend is also found in the case of the photocurrent. In the composite sample, the insulating layer of SiO2 on the Fe3O4 microspheres is the reason for the decrease in the dark current as well as the photocurrent.
 | ||
| Fig. 7 I–V characteristics of (a) pristine Fe3O4 (inset image shows the device structure for the photocurrent measurement of the samples) and (b) Fe3O4@SiO2. | ||
 | ||
| Fig. 8 Photocurrent rise-decay of (a) pristine Fe3O4 and (b) Fe3O4@SiO2 core–shell samples. | ||
To further investigate the role of the surface states/defects during UV On/Off conditions, the time resolved rise and decay of the photocurrent has been measured for both the samples keeping the bias voltage fixed at 10 V. The transient photocurrent response under the steady state illumination of both samples is shown in Fig. 8a and b. Before the transient measurements, the samples were kept in the dark until the current reached an equilibrium value. For the pristine Fe3O4 sample, when UV light is switched on, the photocurrent rises up to 90% of its peak value within ∼400 s and then attains its peak value and gets saturated. When the light is switched off, the photocurrent decays up to 90% within 95 s. In case of the composite sample, when the UV light is switched on, the photocurrent rises up to 90% of its peak value within 105 s. After attaining a peak value, it starts decaying slowly even during steady illumination and eventually reaches a value lower than the value of the dark-current, which may be attributed to carrier relaxation on the SiO2 coating. This observation is similar to the negative photoconductivity observed by S. Panigrahi et al. in ZnO particles embedded in a SiO2 matrix.14 This negative photoconductivity may be a common phenomenon in the composite of SiO2 and transition metal oxide systems. The detailed mechanism of the transient response for both samples under dark and UV illumination is discussed elaborately below.
It is evident from Fig. 8a and b that initially the field dependent dark current starts decreasing slowly until it achieves a steady value (marked by a blue arrow in both Fig. 8a and b), which may be attributed to the field induced adsorption of oxygen molecules as well as to the presence of defects.27 In the absence of UV light, oxygen is adsorbed by taking a free electron from the surface of the Fe3O4 nanoballs. The adsorbed oxygen molecules (O2) on the surface of both pristine Fe3O4 and the Fe3O4@SiO2 core–shell particles become negatively charged ions [O2 + e− → O2−(ad)] after capturing the free electrons from both Fe3O4 and the Fe3O4@SiO2 core–shell particles and develops a depletion layer near the film surface of low conductivity.
Thus, the surface of both type of particles are almost depleted of carriers, leading to low conductivity in the dark.28–30 Under UV illumination, the photogenerated electron-holes are produced and the captured species (O2− ion) are released by the process O2−(ad) + h+ → O2 (g). The adsorbed oxygen molecules are released in air, which lowers the barrier height for the electrons in the Fe3O4 sample. This mechanism was proposed by Muraoka et al.31 When all photoinduced holes react with O2−, the photo current gets saturated in the Fe3O4 samples. Furthermore, it is seen that the photocurrent of Fe3O4@SiO2 is reduced significantly when compared to that of pristine Fe3O4 microspheres. A decrease of photoconductivity has been observed for ZnO quantum dots embedded in a SiO2 matrix,14 Si nanocrystals in SiO2 (ref. 32) and CdSe embedded in SiO2.33 In the Fe3O4@SiO2 core–shell structure, the surface properties of the Fe3O4 microspheres are quite different because of the formation of a long chain SiO2 network around the Fe3O4 core particles. The lower photocurrent observed in the case of the Fe3O4@SiO2 sample is explained with the help of a schematic diagram shown in Fig. 9.
 | ||
| Fig. 9 Schematic representation of the mechanism of anomalous photoconductivity exhibited by Fe3O4@SiO2. | ||
The lower photocurrent observed in the Fe3O4@SiO2 sample when compared to that in pristine Fe3O4 sample can be explained on the basis of adsorption/desorption processes on the surface of the samples. The concentration of adsorbed oxygen molecules on the Fe3O4 surface depends upon the concentration of dangling bonds on the surface of the sample, and as the surface to volume ratio in nano Fe3O4 is large, the surface phenomena of adsorption/desorption plays a significant role. Oxygen molecules get adsorbed on the surface of Fe3O4, and when the surface is illuminated by UV-vis light, photogenerated holes release the adsorbed oxygen from the surface of Fe3O4 and the photoinduced electrons gives rise to the photocurrent in the pristine Fe3O4 sample. In the case of the Fe3O4@SiO2 core–shell sample, a significant proportion of the surface of Fe3O4 nanoparticles is passivated with the SiO2 layer (passivation of such Fe3O4 nanoparticles is shown by black lines in Fig. 9) that results in the reduction of adsorption sites for O2 molecules.
The photocurrent in the core shell may be attributed to the desorption of O2 molecules from the surface of the SiO2 layer as well as from the surfaces of the non-passivated Fe3O4 nanoparticles. As the SiO2 layer has a thickness in the range of 25–30 nm, which is lower than the skin depth for SiO2 in the UV region, a small portion of UV illumination (high energy photons) penetrates the shell and reaches the core to interact with the non-passivated Fe3O4 nanoparticles, and because the surface to volume ratio gets reduced in the core–shell structure when compared to that in the pristine Fe3O4 nanoparticles, photocurrent as a result of desorption of O2 molecules in core–shell nanostructure gets reduced when compared to that in the pristine Fe3O4 nanoparticles. A few electrons tunneling through the SiO2 layer help O2 to get adsorbed on the Fe3O4 surface under dark conditions, whereas upon UV illumination, the photogenerated holes release O2 molecules, which cannot come out from the core into the atmosphere. As a result, the electrons are accumulated on the Fe3O4–SiO2 interface. Anomalous behavior of photocurrent in the core–shell nanostructure may be attributed to the re-adsorption of the desorbed O2 molecules (after getting accumulated electrons from the Fe3O4–SiO2 interface) on the surface of the non-passivated Fe3O4 nanoparticles. Therefore, because of the adsorption and desorption mechanism of oxygen occurring simultaneously under continuous illumination, we observe an anomalous drop in the photocurrent giving rise to the negative PC. After the UV is switched OFF, the decay current follows the oxygen adsorption mechanism.34,35 Thus, when the illumination is terminated, the current reduces faster due to the fast recombination of the electrons and holes.
(4) Conclusions
In summary, we could successfully encapsulate Fe3O4 microspheres made up of nanoparticles with a SiO2 shell. We observed enhanced PL and negative PC for the Fe3O4@SiO2 core@shell material. The Fe3O4 microspheres exhibited a reduced PL intensity and enhanced PC. The enhanced PL emission in the Fe3O4@SiO2 samples is attributed to the reduction of the non-radiative trap levels at the interfaces of the smaller nanoballs after SiO2 coating. SiO2 shell formation reduces the oxygen adsorption sites and the tunneling of electrons among the particles leads to anomalous behavior of negative photoconductivity in Fe3O4@SiO2. To conclude, pristine Fe3O4 can be utilized for UV photodetection and optical switches, whereas the Fe3O4@SiO2 core@shell samples might be used for luminescent materials due to their enhanced PL intensity.Acknowledgements
The authors would like to thank Mr Souvik Banerjee (scientific assistant) and Mr Kousik Bagani (senior research fellow of SINP) for SEM measurements of all the samples. The author Dr Sheo K. Mishra is also grateful to UGC, India, for providing Dr D. S. Kothari's Post-Doctoral Fellowship [no. F. 4-2/2006 (BSR)/13-764/2013 (BSR)].References
- Q. H. Lu, K. L. Yao, D. Xi, Z. I. Liu, X. P. Luo and Q. Ning, Nanoscience, 2006, 11, 241–248 CAS.
- G. H. Du, Z. L. Liu, X. Xia, Q. Chu, S. M. Zhang and J. Sol-Gel, Sci. Technol., 2006, 39, 285–291 CAS.
- S. Banerjee, S. O. Raja, M. Sardar, N. Gayathri, B. Ghosh and A. Dasgupta, J. Appl. Phys., 2011, 109, 123902–123907 CrossRef PubMed.
- Z. L. Liu, Y. J. Liu, K. L. Yao, Z. H. Ding, J. Tao and X. Wang, J. Mater. Synth. Process., 2002, 10, 83–87 CrossRef CAS.
- E. Girgis, M. M. Wahsh, A. G. Othman, L. Bandhu and K. V. Rao, Nanoscale Res. Lett., 2011, 6, 460–467 CrossRef PubMed.
- J. Li, L. Gao, Q. Zhang, R. Feng, H. Xu, J. Wang, D. Sun and C. Xue, J. Nanomater., 2014, 2014, 1–7 Search PubMed.
- W. Wu, X. H. Xiao and S. F. Zhang, Nanoscale Res. Lett., 2011, 6, 533–538 CrossRef PubMed.
- K. Vanheusden, W. L. Warren, C. H. Seager, D. R. Tallant, J. A. Voigt and B. E. Gnade, J. Appl. Phys., 1996, 79, 7983–7990 CrossRef CAS PubMed.
- K. Vanheusden, C. H. Seager, W. L. Warren, D. R. Tallant and J. A. Voigt, Appl. Phys. Lett., 1996, 68, 403–405 CrossRef CAS PubMed.
- Y. Gong, T. Andelman, G. F. Neumark, S. O'Brien and I. L. Kuskovsky, Nanoscale Res. Lett., 2007, 2, 297–302 CrossRef CAS.
- S. Liang, M. Li, J. H. Wang, X. L. Liu, Z. H. Hao, L. Zhou, X. F. Yu and Q. Q. Wang, Opt. Express, 2013, 21, 3253–3258 CrossRef CAS PubMed.
- S. Panigrahi and D. Basak, Chem. Phys. Lett., 2011, 511, 91–96 CrossRef CAS PubMed.
- C. Jin, H. Kim, S. Park, Y. Kwon, S. Lee, H. W. Kim and C. Lee, Adv. Appl. Ceram., 2011, 110, 270–274 CrossRef CAS PubMed.
- S. Panigrahi, A. Bera and D. Basak, ACS Appl. Mater. Interfaces, 2009, 1, 2408–2411 CAS.
- J. D. Prades, F. H. Ramirez, R. J. Diaz, M. Manzanares, T. Andreu, A. Cirera, A. R. Rodriguez and J. R. Morante, Nanotechnology, 2008, 19, 465501–465507 CrossRef CAS PubMed.
- Q. H. Li, T. Gao, Y. G. Wang and T. H. Wang, Appl. Phys. Lett., 2006, 86, 123117–123119 CrossRef PubMed.
- Y. Lin, D. Wang, Q. Zhao, Z. Li, Y. Ma and M. Yang, Nanotechnology, 2006, 17, 2110–2115 CrossRef CAS.
- X. L. Fang, C. Chen, M. S. Jin, Q. Kuang, Z. X. Xie, S. Y. Xie, R. B. Huang and L. S. Zheng, J. Mater. Chem., 2009, 19, 6154–6160 RSC.
- Y. H. Jin, S. D. Seo, H. W. Shim, K. S. Park and D. W. Kim, Nanotechnology, 2012, 23, 125402–125407 CrossRef PubMed.
- B. Sahoo, K. S. P. Devi, S. K. Sahu, S. Nayak, T. K. Maiti, D. Dhara and P. Pramanik, Biomater. Sci., 2013, 1, 647–657 RSC.
- F. Ahangaran, A. Hassanzadeh and S. Nouri, Int. Nano Lett., 2013, 3, 23–27 CrossRef.
- C. Hui, C. Shen, J. Tian, L. Bao, H. Ding, C. Li, Y. Tian, X. Shi and H. J. Gao, Nanoscale, 2011, 3, 701–705 RSC.
- Y. Su, G. Li, X. Bo and L. Li, Nanotechnology, 2007, 18, 485602 CrossRef.
- S. K. Mishra, R. K. Srivastava and S. G. Prakash, J. Alloys Compd., 2012, 539, 1–6 CrossRef CAS PubMed.
- Z.-M. Liao, Z.-K. Lv, Y.-B. Zhou, J. Xu, J.-M. Zhang and D.-P. Yu, Nanotechnology, 2008, 19, 335204 CrossRef PubMed.
- P. K. C. Pillai, N. Shroff, N. N. Kumar and A. K. Tripathi, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 32, 8228–8233 CrossRef CAS.
- S. K. Mishra, R. K. Srivastava, S. G. Prakash, R. S. Yadav and A. C. Pandey, Electron. Mater. Lett., 2011, 7, 31–38 CrossRef CAS PubMed.
- Q. H. Li, T. Gao and T. H. Wang, Appl. Phys. Lett., 2005, 86, 193109–193111 CrossRef PubMed.
- T. Gao, Q. H. Li and T. H. Wang, Appl. Phys. Lett., 2005, 86, 173105–173107 CrossRef PubMed.
- H. Kind, H. Yan, B. Messer, M. Law and P. Yang, Adv. Mater., 2002, 14, 158–160 CrossRef CAS.
- Y. Muraoka, N. Takubo and Z. Hiroi, J. Appl. Phys., 2009, 105, 103702–103708 CrossRef PubMed.
- S. H. Choi and R. G. Elliman, Appl. Phys. Lett., 1999, 74, 3987–3989 CrossRef CAS PubMed.
- E. A. Kafadryan, S. Levichev, S. R. C. Pinto, N. R. Aghmalyanl, R. K. Hovsepyanl, G. R. Badalyan, A. Chahboun, A. G. Rolo and M. J. M. Gomes, Semicond. Sci. Technol., 2008, 23, 095025–095030 CrossRef.
- Q. H. Li, T. Gao, Y. G. Wang and T. H. Wang, Appl. Phys. Lett., 2005, 86, 123117–123119 CrossRef PubMed.
- X. G. Zheng, Q. Sh. Li, W. Hu, D. Chen, N. Zhang, M. J. Shi, J. J. Wang and L. Ch. Zhang, J. Lumin., 2007, 122, 198–201 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |