
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aromatic N-oxide templates open inclusion and dimeric capsular assemblies with methylresorcinarene†
Ngong Kodiah
Beyeh
*,
Rakesh
Puttreddy
and
Kari
Rissanen
*
Department of Chemistry, University of Jyväskylä, P. O. Box 35, 40014, Finland. E-mail: ngong.k.beyeh@jyu.fi; kari.t.rissanen@jyu.fi
First published on 16th March 2015
Abstract
C2-2-methylresorcinarene forms host–guest complexes with pyridine N-oxide and quinoline N-oxide. In solution the NMR studies support the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 host–guest complexes while in the solid state, the single crystal X-ray diffraction studies reveal dimeric capsule-like assemblies with 2:3 and 2:2 host–guest stoichiometry.
:1 host–guest complexes while in the solid state, the single crystal X-ray diffraction studies reveal dimeric capsule-like assemblies with 2:3 and 2:2 host–guest stoichiometry.
Introduction
Resorcinarenes1 constitute a widely studied group of aromatic macrocyclic host compounds. In the C4v conformation, they possess a bowl-shape π-rich cavity suitable for the recognition of many guests through multiple weak interactions. Due to their π-rich cavity, the majority of the reported host–guest complexes with resorcinarenes have been with quaternary ammonium and phosphonium cations.2 These tetrahedral guests interact with the resorcinarene cavity mainly through cation⋯π interactions. Depending on their size and charge distribution, assemblies ranging from open inclusion complexes,3 dimeric4 and hexameric5 assemblies and nanotubes6 have been reported with quaternary ammonium cations. Host–guest complexes involving primary, secondary and tertiary ammonium salts have also been reported.7 The anions predominantly interact either with the hydroxyl groups or through weak C–H⋯anion interactions at the lower rims of the resorcinarenes.2–4Resorcinarenes are also known to host N-heteroaromatic five- and six-membered planar guests, such as imidazole, triazole, pyridine and pyrazine.8 The larger, 10-membered quinoline is usually too large for inclusion, however, the inclusion of 2-pyridylmethanol indicates that the cavity can interact with substituted heterocyclic molecules containing hydroxyl groups.8 Elongated N-aromatic compounds such as 2,2′- and 4,4′-bipyridines as well as caffeine have been shown to co-crystallize with resorcinarenes resulting in different types of architectures.9
The size and electronic properties of pyridine N-oxides9,10 makes this family of compounds important targets to understand their self-assembly processes in supramolecular chemistry. Industrially, these compounds are well credited as oxygen atom transfer reagents in catalysis and routinely used in the syntheses of high-valent transition metal, lanthanide and actinide oxo-complexes.11 The N–O functionality in pyridine N-oxides constitutes a unique property which can act either as electron donor or acceptor group, and as a result makes them potential guest molecules for resorcinarenes. These properties are important and have chemical consequences:
(a) Their π-deficient aromatic rings can interact strongly with electron rich resorcinarenes through suitable π⋯π interactions.12
(b) The lone pairs on N–O groups can function as efficient acceptor10 for multiple hydrogen bonding to the hydroxyl groups of the resorcinarenes.
While there are several reports of interactions between pyridine N-oxides with calixarenes13 and cavitands,14 corresponding studies with core resorcinarenes are unreported.
In this contribution, we study the complexation between C2-2-methylresorcinarene 1 as the receptor, pyridine N-oxide 2 and quinoline N-oxide 3 as guests (Fig. 1). The intercapsular interactions of these N-oxides in the final host–guest assemblies are analysed in solution through 1H NMR spectroscopic studies. The single crystal X-ray diffraction analyses show two supramolecular 2:3 and 2:2 host–guest dimeric capsular assemblies of resorcinarenes 1 as the receptor, with the N-oxides 2 and 3 as included guests.
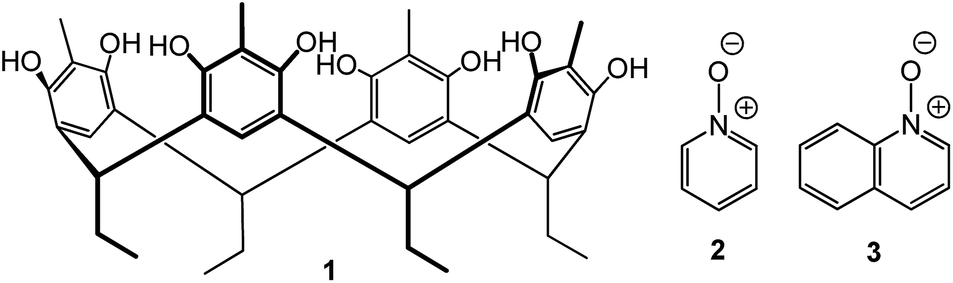 | ||
| Fig. 1 C2-2-methylresorcinarene host 1, pyridine N-oxide 2 and quinoline N-oxide 3 as guests. | ||
Results and discussion
NMR spectroscopy
Host–guest complexes formed between resorcinarenes and tetramethyl ammonium (TMA) salts with the cation sitting in the center of the cavity of the host reveals strong cation⋯π and weak C–H⋯π interactions.2–6 With aromatic N-oxides, the expected binding modes will be π⋯π interactions between the π-rich resorcinarene cavity and the π-poor N-oxide ring, and the accustomed hydrogen bonds between the O atom of the N-oxides and the hydroxyl groups of the resorcinarene. A suitable scenario for maximizing both binding modes between the resorcinarene host 1 and the aromatic N-oxide guests 2 and 3, would be for the guests to sit deep in the resorcinarene cavity with the N–O group pointing upwards.Complexation studies between the C2-2-methylresorcinarene host 1 with the pyridine and quinoline N-oxide guests 2 and 3, were done in CD3OD at 298 K through a series of 1H NMR experiments. Several mixtures of the host 1 and different equivalents of the pyridine N-oxide guest 2 (up to six equivalents) were prepared and the 1H NMR recorded and analyzed (Fig. 2). The observed up field shifts result from the shielding effects of the aromatic rings of the bowl-shaped host cavity upon complexation of the guest 2. This clearly points to a guest exchange fast on the NMR time scale at 298 K. A close look at the 1:1 mixture between the resorcinarene and the pyridine N-oxide 2 shows the para (c) protons to be the most shielded with a shift change of 0.62 ppm and the ortho (a) protons as least shielded with a shift change of 0.25 ppm. These shift changes clearly confirms the orientation of the pyridine N-oxide in the resorcinarene cavity. The large shift change for the para protons suggests that the proton is situated deep in the cavity of the host. Additionally, the breathing effect (Fig. S1a†) of the host through the downfield shift of the host aromatic protons (1:1 mixture, 0.02 ppm) and up field shift of the host methyl protons (1:1 mixture, 0.02 ppm) clearly supports guest binding (Fig. 2 and S1†).
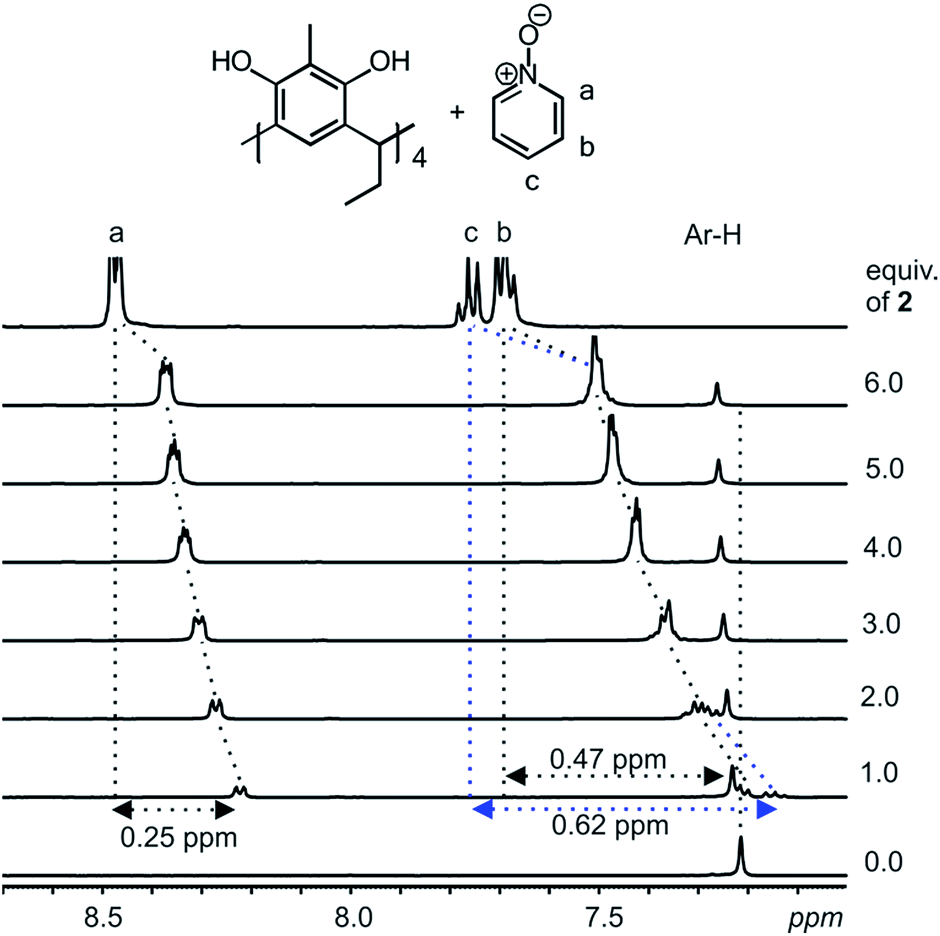 | ||
| Fig. 2
1H NMR (CD3OD, 298 K) spectral changes observed from a mixture of C2-2-methylresorcinarene host 1 and several equivalents of pyridine N-oxide 2. The shift changes of the guest signals in ppm from a 1:1 host–guest mixture are highlighted. Vertical dotted lines gives an indication of the complexation induced shift changes. | ||
With a clear confirmation of the binding of pyridine N-oxide 2 by the C2-2-methylresorcinarene 1, we proceeded to investigate the effect of the size of the aromatic N-oxide in the host–guest process. Accordingly, quinoline N-oxide 3, benzene fused analogue to pyridine N-oxide was selected to probe the effect of size and its 1H NMR shifts caused by the electron delocalization. Several mixtures of the host 1 and different equivalents of the quinoline N-oxide guest 3 (up to six equivalents) were prepared and the 1H NMR was recorded and analyzed (Fig. 3). Clear up field shifts related to the quinoline N-oxide signals are observed, thus confirming the formation of a host–guest complex. A closer look at the 1:1 mixture between the resorcinarene and the quinoline N-oxide 3 will ascertain the orientation of the larger guest in the host cavity. The largest shift was observed with the guest (e) signals (0.67 ppm) while the shift change of the (g) protons was 0.37 ppm. The shift change of the (a) protons (0.15 ppm) was significantly lower when compared with the (e) signals and thus confirm the orientation of the guest 3 in the host 1 cavity. Moreover, the breathing effect (Fig. S1b†) of the host through the downfield shift of the host aromatic protons (1:1 mixture, 0.03 ppm) and up field shift of the host methyl protons (1:1 mixture, 0.08 ppm) where larger than the values observed upon the complexation of the smaller pyridine N-oxide guest 2 (Fig. 2, 3 and S6, S7†).
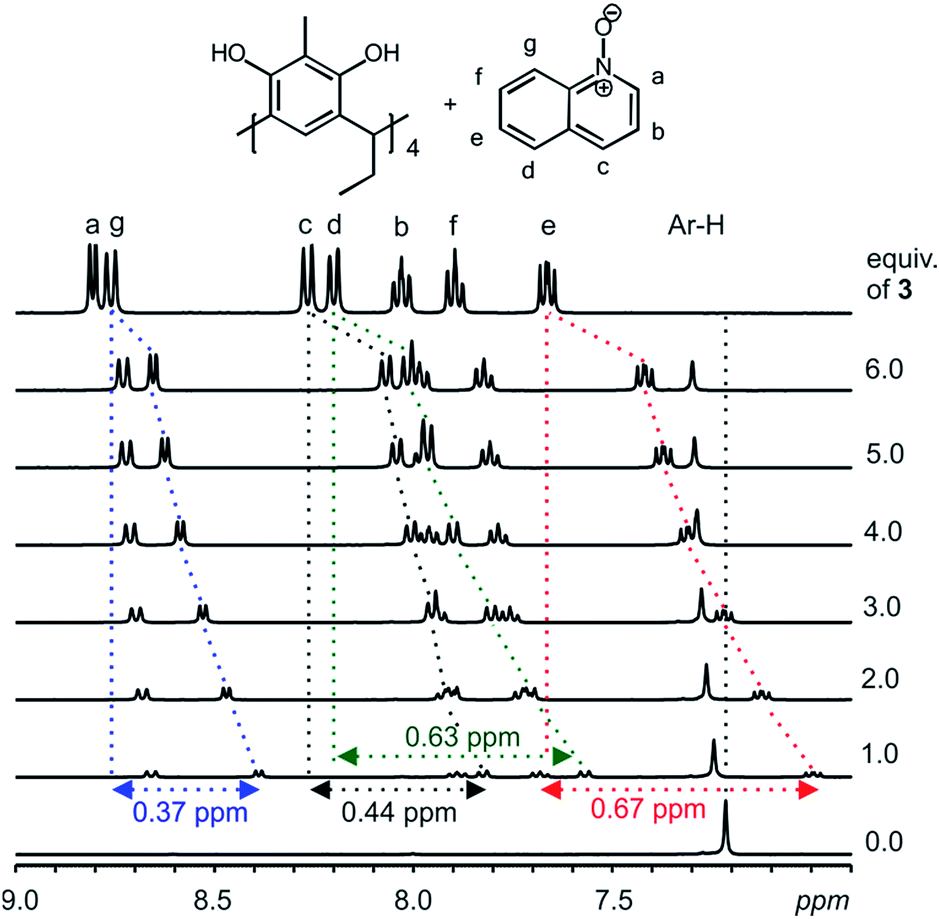 | ||
| Fig. 3
1H NMR (CD3OD, 298 K) spectral changes observed from a mixture of C2-2-methylresorcinarene host 1 and several equivalents of quinoline N-oxide 3. The shift changes of selected guest signals in ppm from a 1:1 host–guest mixture are highlighted. Vertical dotted lines gives an indication of the complexation induced shift changes. | ||
To quantify the binding process between the host and the guests, a series of 1H NMR titration experiments in CD3OD at 298 K were done. For solubility reasons of the host 1, less polar and less competitive solvents could not be used in the titration experiments. Despite the competitive effect of the bulk methanol, significant complexation induced up field shifts of the guests signals were observed. Job plot experiments15 of the host and guests signals indicate a clear 1:1 complex stoichiometry (Fig. S4 and S5†). The lack of formation of larger complexes such as dimeric and/or hexameric assemblies in solution is probably due to the efficiency with which the solvent competes with the hydrogen bonding system of the host especially in bulk amounts.
In the 1H NMR titration experiment, increasing amounts of each of the guests 2 and 3 (200 mM) were added to a solution of the host 1 (10 mM). Signals from both the host and guests were successfully determined and the binding constants were obtained by non-linear least square titration curve fitting of the respective titration data based on 1:1 host–guest binding mode using the HypNMR2008 computer program.16 The association constant for the binding of the quinoline N-oxide 3 (logK = 1.8157 ± 0.0171) was slightly larger than the association constant for the binding of pyridine N-oxide 2 (logK = 1.7527 ± 0.0125). The slightly higher affinity of the host towards the quinoline N-oxide 3 could be explained by the large size of the guest resulting in a better host–guest fit and thus enhanced π⋯π interactions. The generally low binding constant highlights the well known3,4 interference of the bulk methanol.
X-ray crystallography
Single crystal analysis of C2-2-methylresorcinarene host 1 with the guest pyridine N-oxide 2 reveals a dimeric capsule, 23@12(CH3OH)2, constituting a 2:3 host–guest ratio, as shown in Fig. 4a. The dimeric capsule nicely ensembles with two pyridine N-oxide guests accommodated in each host 1 at a depth of ca. 2.87 Å (Fig. S1c†) with N–O group pointing up. As a consequence of such orientation, the pyridine N-oxide orients its para-hydrogen (Fig. 4) towards the aromatic ring of host 1 at distances of ca. 2.90 Å (<C–H⋯π 131°), due to C–H⋯π interaction. Although the meta-hydrogens (Fig. 4) have same distance (d = 2.88 and 3.12 Å, <C–H⋯π 118° and 130°, respectively) to the aromatic rings (Fig. S1c†), it is clear that the orientation and C–H⋯π interactions play a major role. The C–H⋯π interaction for the para-hydrogen in guest 2 indices the chemical shift observed in the 1H NMR spectrum.
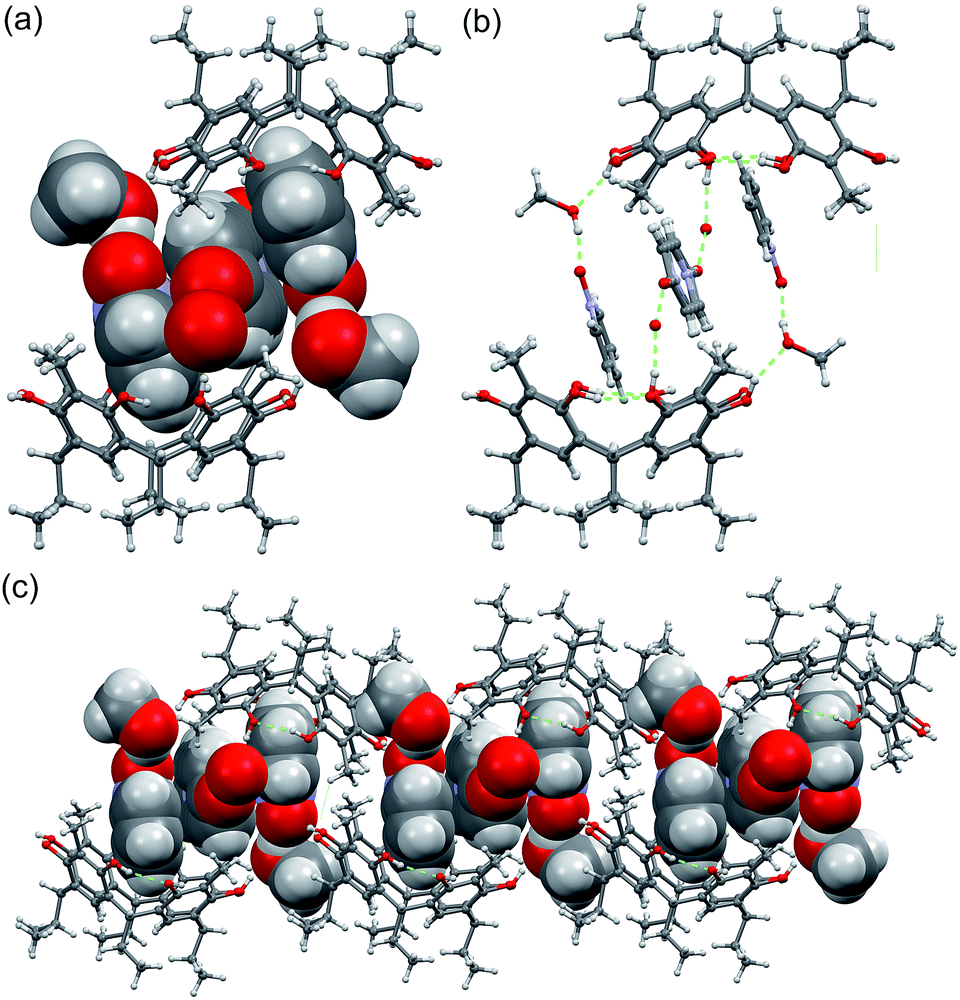 | ||
| Fig. 4 (a) The X-ray structure of 23@12(CH3OH)2 with guests and solvents shown in CPK model. (b) The methanol hydrogen bonding between the dimer halves and (c) packing of the dimers along b-axis. | ||
The N–O groups which point out from the cavity are hydrogen bonded to one methanol molecule each (Fig. 4a), both of which are hydrogen bonded to the hydroxyl group of the capsules resorcinarene half (Fig. 4b), being an essential feature for closing the dimer. The two pyridine N-oxides hosted by the cavity exhibits π–π interaction with the third central pyridine N-oxide with a distance of ca. 3.61 Å. This central pyridine N-oxide is oriented orthogonally and hydrogen bonded to disordered water molecules located outside of the cavity. Such extended π–π interactions between three guest molecules stabilizing a dimeric capsule have not been previously observed in resorcinarene host systems. As a result, the height of the dimer, measured from the centroid-to-centroid distance calculated from the bridging methane carbon atoms, 13.68 Å, and the pseudo-capsular dimer has a staggered conformation with respect to the dimer halves (Fig. S2a†).
In spite of the hydrogen bonding competition between the host, pyridine N-oxide and the protic solvent, the electron rich cavity of resorcinarene retains its C4v conformation stabilized by four –OH⋯H intramolecular hydrogen bond interactions.
The quinoline N-oxide 3, the naphthalene analogue of pyridine N-oxide, under the same complexation conditions with host 1 results in a different pseudo-capsular dimer, 32@12(CH3OH)6 with 2:2 host–guest ratio (Fig. 5.) The centrosymmetric dimer consists of two resorcinarenes, two quinoline N-oxides and six methanol molecules. The quinoline N-oxides are fully encapsulated by the two resorcinarenes and the dimer is closed by the six methanol molecules similarly as in 23@12(CH3OH)2. Two of the six methanol molecules H-bond to 1 from outside (Fig. 5c, yellow color), while the remaining four connect the two dimer halves via the hydroxyl groups of resorcinarene (Fig. 5b).
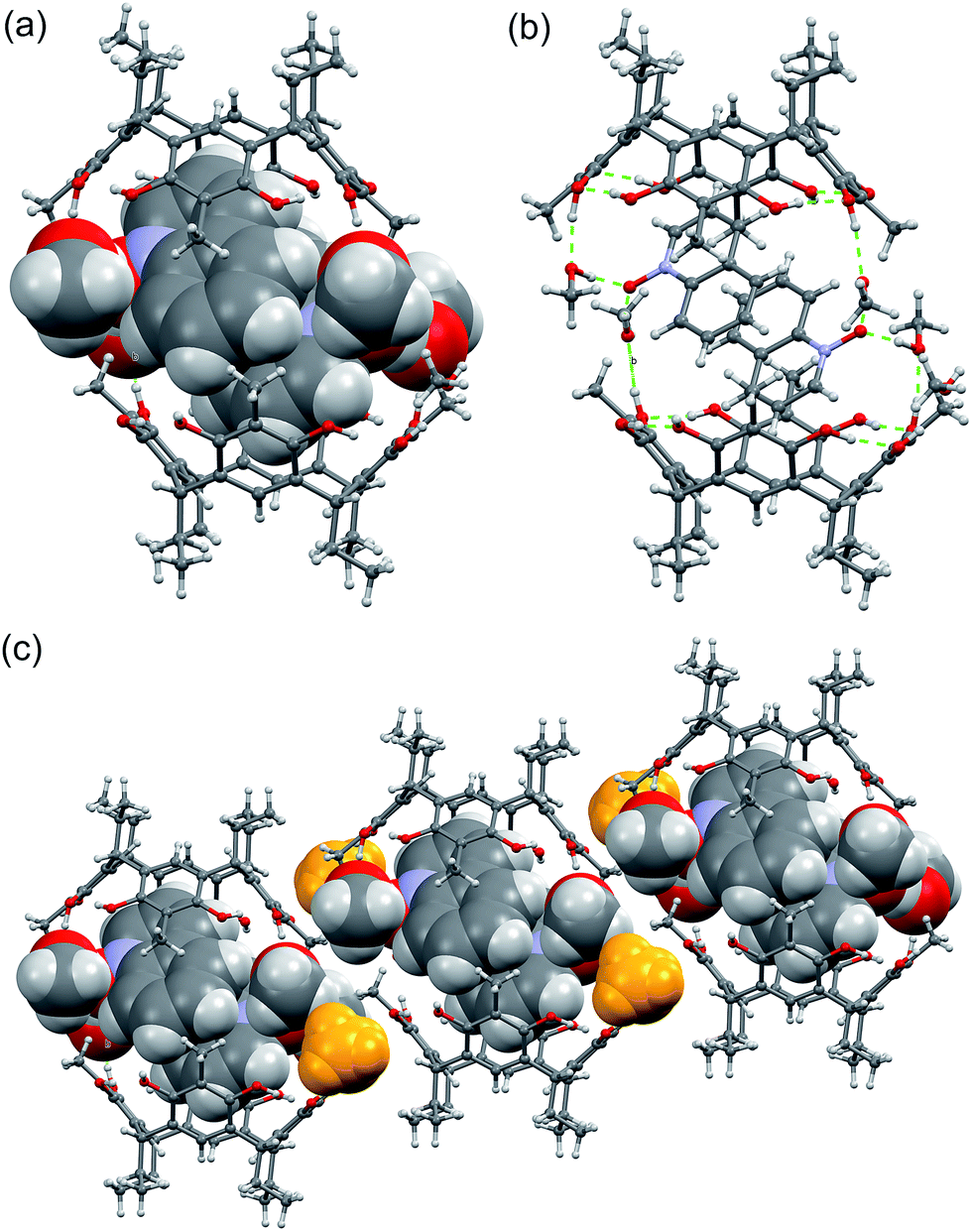 | ||
| Fig. 5 (a) The X-ray structure of 32@12(CH3OH)6 with guests and solvents shown in CPK model. (b) The methanol hydrogen bonding between the dimer halves, and (c) packing of the dimers along b-axis. | ||
The two quinoline N-oxides sit tightly inside the cavity ca. 2.78 Å (Fig. S1d†) above the plane calculated from bridging methane carbon atoms and simultaneously exhibits C–H⋯π interactions with the resorcinarene aromatic rings at distances of around 2.70 Å (<C–H⋯π(centroid) 163°). Furthermore, the dimer is stabilized by the mutual π⋯π interactions between the quinoline N-oxide molecules with centroid-to-centroid distances of 3.56 and 3.67 Å. As the quinoline N-oxides are closely packed inside the dimer cavity, the chemical environment will induce chemical shift changes both to the guest as well as to the host. Although, the hydrogens at the g- and a-position (Fig. 3) are known for C–H functionalization in organic syntheses, in the current study, correlating the observed 1H NMR chemical shift changes with solid state analysis, the larger shift for c-protons (Fig. 3) can be accounted for by the C–H⋯π interactions (Fig. S1d†). At the same time, the chemical shifts for e- and d-hydrogens can be explained by the increased electron deficiency created by the C–H⋯π interactions between the guest and host aromatic systems, supplemented by the π⋯π interactions between the quinoline N-oxide molecules.
In 32@12(CH3OH)6, like in 23@12(CH3OH)2, the co-crystallised methanol molecules play a very important role in dimerization by blocking the “outside” interactions, such as hydrogen bonds, and thus isolating the dimers from each other. Due to the 2:2 host–guest ratio, even quinoline N-oxide being larger in size than pyridine N-oxide, the tight encapsulation of the π⋯π bonded quinoline N-oxide pair into the cavity, reinforced by the methanolic hydrogen bonds, causes a dramatic change in dimer dimensions. The height of the dimer reduces to 11.83 Å with nearly completely eclipsed overall conformation of the dimer halves (Fig. S2b†) resulting is a much tighter dimeric capsule structure.
Conclusions
The work shows the versatility of resorcinarenes in the complexation of a variety of guest compounds utilizing weak interactions. Although there are numerous reports of the binding of ammonium salts by resorcinarenes through the much stronger cation⋯π interactions, limited attention had been invested on the utilization of π⋯π interactions in guest binding. In aromatic N-oxides, both π⋯π and C–H⋯π interactions work in tandem to form host–guest complexes. Slight preference was shown for quinoline N-oxide 3 over the pyridine N-oxide 2 by virtue of better-fit. The interference of the bulk methanol was highlighted in solutions by lack of formation of larger host–guest assemblies other than 1:1 with low binding constants. The X-ray analysis proves resorcinarenes as a remarkable host for N-oxides to template capsule formation by utilizing hydrogen bonding with solvents equally playing important role. Additionally, the breathing nature and spatial orientation of resorcinarene adapting different conformations with different molecules also facilitate the capsule formation. The current investigation shows the way molecules aggregate in solution state and the relative crystal structure predictions based on the NMR shifts caused by host–guest chemistry. The use of π-rich and π-deficient aromatic systems and the study of their stabilization by non-covalent π–π and C–H⋯π interactions show how these molecules might further be exploited to design compounds of biological importance. The information on coordination modes provided by these N-oxides in the resorcinarene cavity could be used in future studies to direct specific multicomponent reactions.
Acknowledgements
We gratefully acknowledge the Academy of Finland (K.R.: grant no. 265328 and 263256; N.K.B.: grant no. 258653) and the University of Jyväskylä for financial support.Notes and references
- (a) P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS; (b) K. Rissanen, Angew. Chem., Int. Ed., 2005, 44, 3652–3654 CrossRef CAS PubMed.
- (a) J. L. Atwood and A. Szumna, Supramol. Chem., 2002, 2, 479–482 CrossRef CAS; (b) N. K. Beyeh and K. Rissanen, Isr. J. Chem., 2011, 51, 769–780 CrossRef CAS; (c) H. Mansikkamaki, M. Nissinen and K. Rissanen, Chem. Commun., 2002, 1902–1903 RSC; (d) N. K. Beyeh, A. Valkonen and K. Rissanen, Supramol. Chem., 2009, 21, 142–148 CrossRef CAS.
- (a) N. K. Beyeh, D. P. Weimann, L. Kaufmann, C. A. Schalley and K. Rissanen, Chem.–Eur. J., 2012, 18, 5552–5557 CrossRef CAS PubMed; (b) H. Mansikkamäki, M. Nissinen and K. Rissanen, CrystEngComm, 2005, 7, 519–526 RSC; (c) N. Kodiah Beyeh, M. Göth, L. Kaufmann, C. A. Schalley and K. Rissanen, Eur. J. Org. Chem., 2014, 80–85 CrossRef CAS.
- (a) H. Mansikkamäki, M. Nissinen, C. A. Schalley and K. Rissanen, New J. Chem., 2003, 27, 88–97 RSC; (b) H. Mansikkamäki, C. A. Schalley, M. Nissinen and K. Rissanen, New J. Chem., 2005, 29, 116–127 RSC; (c) H. Mansikkamäki, M. Nissinen and K. Rissanen, CrystEngComm, 2005, 7, 519–526 RSC; (d) M. Luostarinen, A. Åhman, M. Nissinen and K. Rissanen, Supramol. Chem., 2004, 16, 505–512 CrossRef CAS.
- (a) L. R. MacGillivray and J. L. Atwood, Nature, 1997, 389, 469–472 CrossRef CAS; (b) N. K. Beyeh, M. Kogej, A. Åhman, K. Rissanen and C. A. Schalley, Angew. Chem., Int. Ed., 2006, 45, 5214–5218 CrossRef CAS PubMed; (c) T. Gerkensmeier, W. Iwanek, C. Agena, R. Fröhlich, S. Kotila, C. Näther and J. Mattay, Eur. J. Org. Chem., 1999, 2257–2262 CrossRef CAS.
- (a) H. Mansikkamäki, M. Nissinen and K. Rissanen, Angew. Chem., Int. Ed., 2004, 43, 1243–1246 CrossRef PubMed; (b) H. Mansikkamäki, S. Busi, M. Nissinen, A. Åhman and K. Rissanen, Chem.–Eur. J., 2006, 12, 4289–4296 CrossRef PubMed.
- (a) D. A. Fowler, J. Tian, C. Barnes, S. J. Teat and J. L. Atwood, CrystEngComm, 2011, 13, 1446–1449 RSC; (b) A. Shivanyuk, K. Rissanen and E. Kolehmainen, Chem. Commun., 2000, 1107–1108 RSC; (c) N. K. Beyeh, A. Valkonen and K. Rissanen, CrystEngComm, 2014, 16, 3758–3764 RSC; (d) A. Shivanyuk, E. F. Paulus, K. Rissanen, E. Kolehmainen and V. Böhmer, Chem.–Eur. J., 2001, 7, 1944–1951 CrossRef CAS PubMed; (e) A. Shivanyuk, J. C. Friese, S. Döring and J. Rebek, J. Org. Chem., 2003, 68, 6489–6496 CrossRef CAS PubMed; (f) P. Thuery and M. Nierlich, Supramol. Chem., 2000, 11, 185–190 CrossRef CAS; (g) I. Fujisawa, N. Harima, K. Takagi, S. Hiraoka, M. Shionoya, K. Murayama, S. Itsuno, R. Kato and K. Aoki, Bull. Chem. Soc. Jpn., 2012, 85, 1037–1045 CrossRef CAS; (h) N. K. Beyeh, F. Pan, A. Valkonen and K. Rissanen, CrystEngComm, 2015, 17, 1182–1188 RSC.
- (a) M. Nissinen, E. Wegelius, D. Falabu and K. Rissanen, CrystEngComm, 2000, 28, 1–3 Search PubMed; (b) M. Nissinen and K. Rissanen, Supramol. Chem., 2003, 15, 581–590 CrossRef CAS.
- (a) B. Q. Ma and P. Coppens, Cryst. Growth Des., 2004, 4, 1377–1385 CrossRef CAS; (b) B. Q. Ma, Y. Zhang and P. Coppens, Cryst. Growth Des., 2001, 1, 271–275 CrossRef CAS; (c) L. R. MacGillivray, P. R. Diamente, J. L. Reid and J. A. Ripmeester, Chem. Commun., 2000, 359–360 RSC; (d) G. Ferguson, C. Glidewell, A. J. Lough, G. D. McManus and P. R. Meehan, J. Mater. Chem., 1998, 8, 2339–2345 RSC; (e) P. Thuery, M. Nierlich, Z. Asfari, J. Vicens, O. Morikawa and H. Konishi, Supramol. Chem., 2001, 13, 521–527 CrossRef CAS; (f) C. L. Raston and G. W. V. Cave, Chem.–Eur. J., 2004, 10, 279–282 CrossRef CAS PubMed; (g) G. W. V. Cave, M. J. Hardie, B. A. Roberts and C. L. Raston, Eur. J. Org. Chem., 2001, 3227–3231 CrossRef CAS; (h) P. O. Brown, G. D. Enright and J. A. Ripmeester, CrystEngComm, 2006, 8, 381–383 RSC.
- (a) G. Caron, G. Ermondi, D. Boschi, P. A. Carrupt, R. Fruttero, B. Testa and A. Gasco, Helv. Chim. Acta, 1999, 82, 1630–1639 CrossRef CAS; (b) C. S. Burgey, K. A. Robinson, T. A. Lyle, P. G. Nantermet, H. G. Selnick, R. C. A. Isaacs, S. D. Lewis, B. J. Lucas, J. A. Krueger, R. Singh, C. Miller-Stein, R. B. White, B. Wong, E. A. Lyle, M. T. Stranieri, J. J. Cook, D. R. McMasters, J. M. Pellicore, S. Pal, A. A. Wallace, F. C. Clayton, D. Bohn, D. C. Welsh, J. J. Lynch, Y. Yan, Z. Chen, L. Kuo, S. J. Gardell, J. A. Shafer and J. P. Vacca, Bioorg. Med. Chem. Lett., 2003, 13, 1353–1357 CrossRef CAS PubMed.
- (a) R. R. Schrock, Chem. Rev., 2001, 102, 145–180 CrossRef; (b) S. M. Mullins, A. P. Duncan, R. G. Bergman and J. Arnold, Inorg. Chem., 2001, 40, 6952–6963 CrossRef CAS PubMed; (c) K.-M. Sung and R. H. Holm, J. Am. Chem. Soc., 2001, 123, 1931–1943 CrossRef CAS PubMed; (d) J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2005, 127, 1338–1339 CrossRef CAS PubMed; (e) D. S. J. Arney and C. J. Burns, J. Am. Chem. Soc., 1995, 117, 9448–9460 CrossRef CAS; (f) J. Jia, A. J. Blake, N. R. Champness, P. Hubberstey, C. Wilson and M. Schröder, Inorg. Chem., 2008, 47, 8652–8664 CrossRef CAS PubMed; (g) A. E. V. Gorden, J. Xu, K. N. Raymond and P. Durbin, Chem. Rev., 2003, 103, 4207–4282 CrossRef CAS PubMed.
- J. W. Steed and J. L. Atwood, in Supramolecular Chemistry, John Wiley & Sons, Ltd, 2009, pp. 591–706 Search PubMed.
- G. Zheng, Y.-Y. Li, H.-D. Guo, S.-Y. Song and H.-J. Zhang, Chem. Commun., 2008, 4918–4920 RSC.
- L. Adriaenssens and P. Ballester, Chem. Soc. Rev., 2013, 42, 3261–3277 RSC.
- (a) K. A. Connors, Binding Constants: The Measurement of Molecular Complex Stability, JohnWiley & Sons, Inc., Canada, 1987 Search PubMed; (b) K. Hirose, J. Inclusion Phenom. Macrocyclic Chem., 2001, 39, 193–209 CrossRef CAS.
- C. Frassineti, S. Ghelli, P. Gans, A. Sabatini, M. S. Moruzzi and A. Vacca, Anal. Biochem., 1995, 231, 374–382 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: X-ray crystallographic and NMR spectroscopic data. CCDC 1051458 and 1051459. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra03667d |
| This journal is © The Royal Society of Chemistry 2015 |