DOI:
10.1039/C5RA03589A
(Paper)
RSC Adv., 2015,
5, 32460-32468
Synthesis and properties of monomer cast nylon-6-b-polyether amine copolymers with different structures
Received
28th February 2015
, Accepted 24th March 2015
First published on 24th March 2015
Abstract
TDI-polyether amine macro-initiators with different structures were synthesized to initiate the caprolactam monomer successfully, and monomer cast (MC) nylon-6-b-polyether amine copolymers with multi-branched molecular structure were prepared via an in situ polymerization. It was found that the apparent activation energy (E) and the pre-exponential factor (A) of the reaction increased for the copolymers, and the TDI-polyether amine macro-initiators presented low reactivity due to the steric effects of the multi-armed long-chain structure. The hydrogen bond and inter-molecular forces of the copolymers were weakened by the introduction of multi-branched molecular chains, which was in favor of the formation of a γ-crystal. The crystallization ability decreased for the copolymers, among which MC nylon-6-b-PP3A with an extra branched-chain showed the highest melting and crystallization temperatures and crystallinity with a relatively large crystal grain size. A pseudo-plastic fluid characteristic of flow behavior was observed for MC nylon-6. The viscoelastic moduli (G) and entanglement density (νe) of all copolymers were higher than that of neat MC nylon-6 due to severe entanglement of the large quantity of multi-branched molecular chains of the copolymers resulting in the formation of physical networks, of which MC nylon-6-b-PP3A presented the highest. In the meantime, the values of the loss factor of the copolymers increased and the storage modulus and glass transition temperature (Tg) decreased. The stress–strain curves of all copolymers showed an untypical yield point and presented obvious strain hardening behavior, while a hairy fibrous structure was observed on the fracture surface of the copolymers, indicating the notable toughening effect of polyether amine on the nylon-6 matrix. The toughening mechanism of the copolymers was deduced as a multi-layer crack extension mechanism.
1. Introduction
Monomer cast nylon-6 is a new type of engineering plastic, which is synthesized by anionic polymerization with caprolactam as the major raw material. Compared with ordinary nylon-6, it has the advantages of high molecular weight, high mechanical strength and excellent self-lubricating performance,1,2 and is widely used to replace metallic materials for the production of gears, bearings etc.3
However, MC nylon-6 has a disadvantage in its low-notched impact strength. It is not resistant to crack propagation, which often results in brittle fracture. The inherent notch sensitivity of MC nylon-6 precludes applications as automotive fenders, body panels etc., which require high impact resistance. Many attempts have been made to improve the toughening properties of MC nylon-6 by blending soft components or adding plasticizing agents, such as ultra-high molecular weight polyethylene (UHMWPE) powders,4 hexamethylphosphoric triamide (HPT)5 etc. Some researchers attempted to toughen MC nylon-6 by preparing novel catalysts or activators with a multi-functional group6 or copolymerized it with other components.7 However, as reported, most of the copolymers with phase or micro-phase separation showed a low tensile strength even though the impact toughness was improved.
Polyether amines with low molecular weights have been widely applied in many fields. They have been used as an important raw material of polyurea elastomers and acted as a hardener of epoxy resins. The flexible structure of the C–O–C bond in polyether amine molecular chains endow the material with excellent toughness, and it’s a good candidate as an organic toughening material. In this work polyether amines with different molecular structures were used for the first time as the copolymerization component in the MC nylon-6 matrix. The active amino groups may react with the isocyanate group of polyisocyanate, and thus the macro-initiator can be prepared for anionic polymerization of caprolactam. The effect of the molecular structure of polyether amine on the reaction kinetics of caprolactam polymerization was studied, the molecular interaction, rheology and crystallization behavior of the copolymers were characterized, and the toughening mechanism was explored.
2. Experimental
2.1 Materials
The caprolactam monomer was supplied by China Petroleum and Chemical Co. Ltd., as a commercial grade product. NaOH (sodium hydroxide) of analytical purity was purchased from Kermel Chemical Reagent Co. Ltd (Tianjin, China), and TDI (toluene-2, 4-diisocyanate) was purchased from Kelong Chemical Reagent Factory (Chengdu, China). Polyether amines with average molecular weights of 2000 g mol−1 (PE2A, PPAD) and 3000 g mol−1 (PP3A) were purchased from Huntsman Polyurethanes Ltd Guangzhou Branch (Guangzhou, China).
2.2 Synthesis and preparation
2.2.1 Synthesis of the TDI-polyether amine macro-initiators. The TDI-polyether amine macro-initiators were prepared at room temperature using a constant temperature bath. A flask was equipped with a magnetic stirrer and charged with 6.96 g TDI. Then 20 g polyether amine was slowly dropped into it. The reaction lasted for 2 h to make sure the reaction was complete. Then the TDI-polyether amine macro-initiators were obtained.
2.2.2 Synthesis of the MC nylon-6-b-polyether amine copolymers. 4 mol caprolactam (CL) was put into a flask and heated to about 130 °C. After completely melting, the melt was refluxed under vacuum for about 30 min to remove any water. Then 0.01 mol NaOH was added under vigorous stirring. The melt was refluxed under vacuum for another 30 min, and then 0.01 mol TDI-polyether amine macro-initiator was added. After quick mixing, the melt was cast into a preheated mould at 160 °C. The reaction lasted for 40 min. The product MC nylon-6-b-polyether amine copolymer was then obtained.
2.3 Measurements
2.3.1 Reaction kinetics analysis. The caprolactam polymerization reaction was conducted in a thermal isolated oven. The reaction temperature during polymerization was measured with a TM-902C thermometer from Shanglong Electric Co. Ltd. (Shanghai, China), and the reaction kinetics were analyzed with the recorded reaction temperature.
2.3.2 Viscosity average molecular weight. The specific viscosity ηsp of neat MC nylon-6 and the copolymers was measured using an Ubbelohde viscometer. Formic acid was used as the solvent for the nylon-6 materials.The intrinsic viscosity is described as the limit of the ratio between the specific viscosity (ηsp) and the polymer concentration (c), as the polymer concentration approaches zero.
| |
[η] = lim![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ηsp/c (c → 0) ηsp/c (c → 0)
| (1) |
By measuring the intrinsic viscosity of polymer solutions, the polymer average molecular weight can be predicted through the Mark–Houwink empirical equation, as described by:
where [
η] is the intrinsic viscosity, and
K and
α are the parameters that depend on the solvent/polymer pair (the values of 22.6 × 10
−3 and 0.82 were chosen respectively).
8 Mη is the viscosity average molecular weight of the polymer.
2.3.3 FTIR analysis. The structure of the neat MC nylon-6 and copolymers was analyzed using a Nicolet-560 Fourier-transform infrared spectrometer (FTIR) (USA). The scanning rate was 20 min−1, and the differentiate rate was 4 cm−1.
2.3.4 Non-isothermal crystallization analysis. The non-isothermal crystallization of neat MC nylon-6 and the copolymers was performed using a Netzsch 204 Phoenix differential scanning calorimeter (DSC) (Germany). The range of the test temperature was 20 °C to 250 °C which was calibrated with indium. The heating rate and cooling rate were 10 °C min−1 under a nitrogen atmosphere. The crystallinity (Xc,DSC) can be calculated using the following equation:| | |
Xc,DSC = [ΔHm/(wΔH0)] × 100%
| (3) |
where w is the mass fraction of each component, ΔHm is the melting enthalpy of the samples and ΔH0 is the balance melting enthalpy, i.e., the melting enthalpy of 100% crystallizing polymer, which is 230 J g−1 for MC nylon-6.9
2.3.5 X-ray diffraction (XRD) analysis. The XRD analysis of neat MC nylon-6 and the copolymers was performed using a Rigaku D/max IIIB X-ray diffractometer (Japan) at room temperature. XRD data were collected from 5 to 40°.
2.3.6 Scanning electron microscopy (SEM) analysis. The morphology of the impact fractured surface of neat MC nylon-6 and the copolymers was observed using a JEOL JSM-5900LV scanning electron microscope (Japan). The operating voltage was 5 kV and the samples were ion beam sputter-coated with gold.
2.3.7 Dynamic rheological analysis. Rheological characterization of neat MC nylon-6 and the copolymers was carried out at 250 °C using a rheometer (TA-AR2000ex, USA) with a 25 mm-parallel plate in a frequency range of 0.01–100 Hz and a 1% strain value. The testing sample disks had a thickness of 1.5 mm and a diameter of 25 mm.
2.3.8 Dynamic mechanical analysis (DMA). The dynamic mechanical analysis of neat MC nylon-6 and the copolymers was carried out using a TA Instrument Q800 (USA). All the samples were measured with a bending mode at a heating rate of 3 °C min−1 and a frequency of 1 Hz. The sample size was 40 × 10 × 4 mm3.
2.3.9 Mechanical properties. The tensile performance of neat MC nylon-6 and the copolymers was measured using a 4302 material testing machine from Instron Co. (USA.) and the notched charpy impact strength was measured using a ZBC-4B impact testing machine from Xinsansi Co. (Shenzhen, China).
3. Results and discussion
3.1 Synthesis and structure of the MC nylon-6-b-polyether amine copolymers
Polyether amines with different structures were applied to react with TDI based on the addition reaction between the amino group of the polyether amines and the isocyanate group of TDI (PE2A with a ethylene oxide main chain and two end amine groups, PP2A with a propylene oxide main chain and two end amine groups and PP3A with a propylene oxide main chain and three end amine groups). Compared with PE2A, the unit structure of PP2A contained an extra methyl side group while PP3A had an extra branched-chain compared with PP2A. The reaction process of multi-armed TDI-polyether amine macro-initiators was as follows:
MC nylon-6-b-polyether amine copolymers were fabricated by means of a cast moulding technique via an anionic ring-opening polymerization mechanism, using NaOH as the catalyst and the above TDI-polyether amines as the macro-initiator.10,11 The reaction formula and 1H NMR analysis of the copolymer was as follows: δHa: 1.58 ppm; δHb: 1.82 ppm, 1.90 ppm, 2.79 ppm; δHc: 3.63 ppm; δHd: 2.23 ppm; δHe: 1.49 ppm.
3.1.1 Reaction kinetics. The temperature–time relationship curves of neat MC nylon-6 and the copolymers during the reaction process are shown in Fig. 1(a). The polymerization process started when the caprolactam melt was cast into a preheated mould. Therefore, at first, the temperature of the reaction system decreased from the mold temperature (160 °C) to the caprolactam melt temperature (130 °C) and then increased gradually with time. Over about 400–1200 s, the temperature of the reaction system tended to be stable, indicating the end of the polymerization process. The reaction temperature of the neat MC nylon-6 which was initiated by TDI increased rapidly and the reaction process terminated in a short time, while that of the copolymer systems increased slowly, indicating a relatively low initiation activity of the TDI-polyether amine macro-initiator. Fig. 1(b) shows that the heating-rate of the copolymers increased first and then decreased over time. By introducing polyether amine, the maximum heating-rate (Rmax) decreased and the time corresponding to the maximum heating-rate (tmax) was significantly prolonged.
 |
| | Fig. 1 Temperature–time relationship curves of the synthesis of MC nylon-6-b-polyether amine copolymers. | |
As to an exothermic chemical reaction, the universal equation for studying the non-isothermal reaction kinetics can be described following:12
| | |
dα/dt = (1 − a)nAe(−E/RT)
| (4) |
where
α is the reaction conversion rate,
t is the reaction time,
n is the reaction order,
A is the pre-exponential factor,
T is the reaction temperature,
E is the apparent activation energy of the reaction and
R is the gas constant with a value of 8.314 J (K
−1 mol
−1).
The polymerization reaction was conducted in a thermal isolated oven. Therefore, the reaction can be considered to be conducted in a homogeneous environment. The reaction conversion rate α can be represented by the increase of the reaction temperature as follows:
| | |
da/dt = 1/(Tf − T0)dT/dt
| (5) |
where
T0,
Tf and
T are the initial reaction temperature, final reaction temperature and reaction temperature at reaction time
t, respectively. Finally,
eqn (6) was obtained after combining
eqn (4) and
(5) as follows:
| | |
ln[1/(Tf − T0)dT/dt] = lnA + nln[1 − (T − T0)/(Tf − T0)] − E/RT.
| (6) |
By calculating eqn (4) at different temperatures, the reaction kinetics parameters can be obtained, as shown in Table 1. The E and A of all copolymers increased, indicating that the polymerization reaction became difficult. The macro-initiator presented low reactivity due to the steric effects of the multi-armed long-chain structure. The reaction order n of neat nylon-6 was approximated to 1, indicating that the copolymerization reaction process was deviated from a one-step reaction. The MC nylon-6-b-PE2A showed the highest E among the copolymers while that of MC nylon-6-b-PP2A and MC nylon-6-b-PP3A was low and of a similar value. The initiation activity of the macro-initiators was affected by the length of the molecular chain when the initiators had the same active center structure. The length of the molecular chain of PP3A was the same as PP2A, which resulted in similar initiation activities of the macro-initiators. The PE2A presented a longer molecular chain and resulted in a lower initiation activity of the macro-initiator.
Table 1 Reaction kinetics parameters of the MC nylon-6-b-polyether amine copolymers
| Sample |
E (kJ mol−1) |
A (s−1) |
n |
Rmax (°C s−1) |
tmax (s) |
| Neat MC nylon-6 |
82.37 |
4.26 × 107 |
0.95 |
0.166 |
295 |
| MC nylon-6-b-PE2A |
131.44 |
1.45 × 1013 |
1.69 |
0.083 |
598 |
| MC nylon-6-b-PP2A |
127.63 |
1.42 × 1013 |
1.62 |
0.087 |
544 |
| MC nylon-6-b-PP3A |
128.34 |
1.39 × 1013 |
1.64 |
0.085 |
551 |
3.1.2 Molecular weight. The molecular weights of neat MC nylon-6 and the copolymers were obtained using a viscosity method and are shown in Table 2. It can be seen that the molecular weight of the copolymers decreased obviously when compared with that of neat MC nylon-6, resulting from the decreasing reactivity and reaction rate of the polymerization process due to the introduction of polyether amine. The MC nylon-6-b-PP3A copolymer presented the highest molecular weight among the copolymers because of its more branched-chain while the MC nylon-6-b-PP2A and MC nylon-6-b-PE2A copolymers showed a similar molecular weight even though their reactivity had a little difference.
Table 2 Viscosity average molecular weight of the MC nylon-6-b-polyether amine copolymers
| Sample |
Neat MC nylon-6 |
MC nylon-6-b-PE2A |
MC nylon-6-b-PP2A |
MC nylon-6-b-PP3A |
| Molecular weight (g mol−1) |
94508.7 |
50648.9 |
48483.8 |
58138.4 |
3.1.3 Molecular interaction. The FTIR spectra analysis for neat MC nylon-6 and the copolymers are shown in Fig. 2. Compared with neat nylon-6, the FTIR peak at 3298.4 cm−1 for the copolymers corresponding to the hydrogen-bonded –NH had no significant change. However, all the copolymers with different molecular structures of the polyether amine showed a new peak at 3499.5 cm−1 which was attributed to non-hydrogen-bonded –NH. Additionally, the absorption peak of amide II’s octave at 3060.4 cm−1 showed varying degrees of blue shift in the copolymers, indicating the increasing number of free amide groups. The results reveal that the amide group in neat nylon-6 almost presented as hydrogen-bonded –NH, while the hydrogen bond and inter-molecular forces of the copolymers were weakened by the introduction of multi-branched molecular chains.
 |
| | Fig. 2 FTIR spectra of the MC nylon-6-b-polyether amine copolymers. | |
Compared with neat MC nylon-6, the blue shift in amide II’s octave of MC nylon-6-b-PP2A was more obvious than that of MC nylon-6-b-PE2A. The major methyl side group in the main chain of PP2A increased the steric hindrance of the molecular chain and further weakened the hydrogen bond. Although the major methyl side group and more branched-chain in PP3A will increase the steric hindrance of the molecular chain, the extra branched-chain can also form hydrogen bonds and partially offset the effect of steric hindrance on the weakened hydrogen bonds.
3.2 Crystallization properties of the MC nylon-6-b-polyether amine copolymers
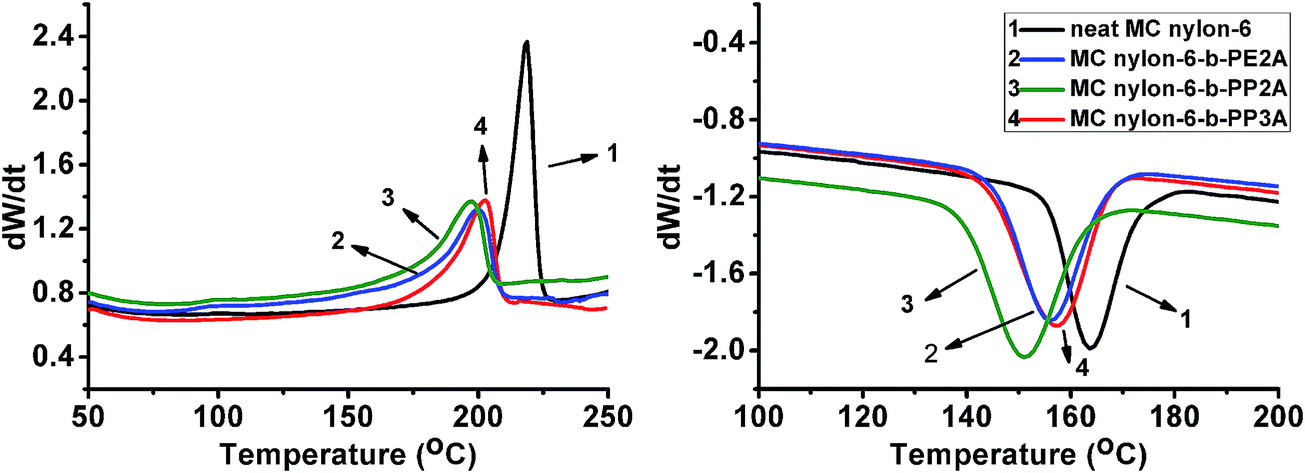
3.2.1 Crystallization behavior. The non-isothermal DSC curves of neat MC nylon-6 and the copolymers are shown in Fig. 3, the melting process presented only one melting peak in the whole temperature range which corresponded to the melting peak of the nylon-6 matrix for all samples, and similar behavior can be observed for the crystallization process, indicating that all the polyether amine components with different molecular structures, incorporated into the nylon-6 main chains, were in the amorphous state. Moreover, it can be seen that the introduction of polyether amine led to an obvious decrease of the melting and crystallization temperatures and the crystallinity of the copolymers compared with that of neat MC nylon-6. The increase of the half peak width indicates the decrease of the crystalline growth rate. Among the copolymers, the MC nylon-6-b-PP3A showed the highest melting and crystallization temperatures and crystallinity. Even though the more branched-chain of nylon-6-b-PP3A will increase the steric hindrance, the extra branched-chain can also crystallize and result in better crystalline properties. The methyl side group in the PP2A molecular chain may lead to a relatively low degree of molecular regularity of the copolymer, which will be unfavorable for the crystallization of the copolymer.
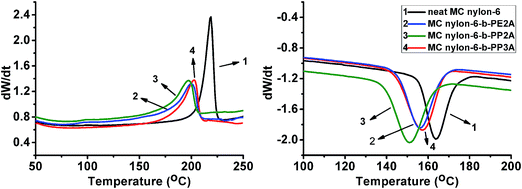 |
| | Fig. 3 Non-isothermal DSC curves of the MC nylon-6-b-polyether amine copolymers. | |
3.2.2 Crystal structure. The crystal structure of neat MC nylon-6 and the copolymers were evaluated using X-ray diffraction (XRD) analysis. As shown in Fig. 4, for all samples, the observed diffractions at 2θ = 20° and 23° corresponded to the α crystalline form of the nylon-6 matrix.13,14 In addition, the MC nylon-6-b-polyether amine copolymers presented a new and small diffraction peak at 2θ = 9.45°, which was attributed to the γ crystalline form. The disordered arrangement of the multi-branched nylon-6 molecular chains was in favor of the formation of the γ-form with long and weak hydrogen bonds. The XRD spectrogram indicates that the crystallization process of the copolymers mainly provides the α-form and a small amount of the γ-form is also formed simultaneously. The crystallinity (Xc,XRD) and crystal grain sizes were calculated with the following expressions, respectively:| | |
Xc,XRD = [Ic/(Ic + Ia)] × 100%, Lhlk = kλ/(βcosθ)
| (7) |
where Ic and Ia are the areas under the crystalline curve and amorphous curve respectively, Lhlk is the size of the crystallites from the normal direction of the hlk plane, k is the Scherrer constant, λ is the wavelength of radiation (equal to 0.154 nm in the present case), β is the full width of half maximum of the diffraction peak (hlk) and θ is the Bragg angle.
 |
| | Fig. 4 XRD spectra of the MC nylon-6-b-polyether amine copolymers. | |
As shown in Table 3, the crystallinity of the nylon-6 matrix decreased with the introduction of the polyether amine component which was coincident with the DSC analysis. The crystal grain size of MC nylon-6-PP2A was relatively small, while MC nylon-6-PP3A with an extra branched-chain had a relatively large crystal grain size.
Table 3 XRD parameters of the MC nylon-6-b-polyether amine block copolymers
| Sample |
Angle (2θ) |
Xc,XRD (%) |
Grain size (Å) |
| α1 (°) |
α2 (°) |
γ (°) |
| Neat Nylon-6 |
20.19 |
23.77 |
|
35.68 |
88 |
| MC nylon-6-b-PE2A |
20.29 |
24.02 |
9.48 |
30.04 |
71 |
| MC nylon-6-b-PP2A |
20.39 |
24.11 |
9.42 |
29.35 |
69 |
| MC nylon-6-b-PP3A |
20.18 |
23.91 |
9.45 |
32.11 |
75 |
3.3 Rheological behavior of the MC nylon-6-b-polyether amine copolymers
The molecular chain entanglement behavior of neat MC nylon-6 and the copolymers was investigated using dynamical rheological measurements. As shown in Fig. 5(a), the complex viscosity as a function of frequency, a pseudo-plastic fluid characteristic of flow behavior was observed for MC nylon-6. On introduction of polyether amine, the copolymers presented a more evident characteristic of shear-thinning when compared to neat MC nylon-6. The difference in complex viscosity between neat MC nylon-6 and the copolymers may be closely related to the difference in the molecular chain structure.
 |
| | Fig. 5 Rheological behavior of the MC nylon-6-b-polyether amine copolymers. | |
The linear viscoelastic moduli (G) of the neat MC nylon-6 and the copolymers are shown in Fig. 5(b and c) as a function of frequency. For all samples, both the elastic modulus and viscous modulus increased with the increase of frequency. When comparing the samples at the same frequency, it can be seen that the viscoelastic moduli of all copolymers were higher than that of neat MC nylon-6. This result was due to the severe entanglement of the large quantity of multi-branched molecular chains through the formation of physical networks in the copolymers. Moreover, among the copolymers, the viscoelastic moduli of MC nylon-6-b-PP3A were highest, which was probably due to it having the most branched molecular chain and severe entanglement. The degree of molecular regularity and crystallinity of MC nylon-6-b-PE2A was slightly higher than that of MC nylon-6-b-PP2A, which led the molecular chain to move in a high shear force.
The entanglement molecular weight (Me) is introduced from the kinetic theory of rubber elasticity. It can be calculated from the plateau modulus (G0N) and can be interpreted as the apparent average molecular weight between coupling junctions as follows:15,16
| |
 | (8) |
The entanglement density νe can be calculated with the following equation:
where
ρ is the polymer density,
R is the gas constant and
T is the absolute temperature.
The Me and νe of the samples are listed in Table 4. It can be seen that with the introduction of polyether amine the νe increased significantly and the Me decreased significantly. The νe of MC nylon-6-b-PP3A was the highest among the copolymers.
Table 4 G0N, νe and Me of the MC nylon-6-b-polyether amine copolymers
| Sample |
G0N (Pa) |
νe (mol m−3) |
Me (g mol−1) |
| Neat MC nylon-6 |
3173.16 |
0.91 |
1228153 |
| MC nylon-6-b-PE2A |
13642.15 |
3.92 |
285668 |
| MC nylon-6-b-PP2A |
7851.16 |
2.25 |
496376 |
| MC nylon-6-b-PP3A |
17442.70 |
5.01 |
223424 |
3.4 Mechanical properties of the MC nylon-6-b-polyether amine copolymers
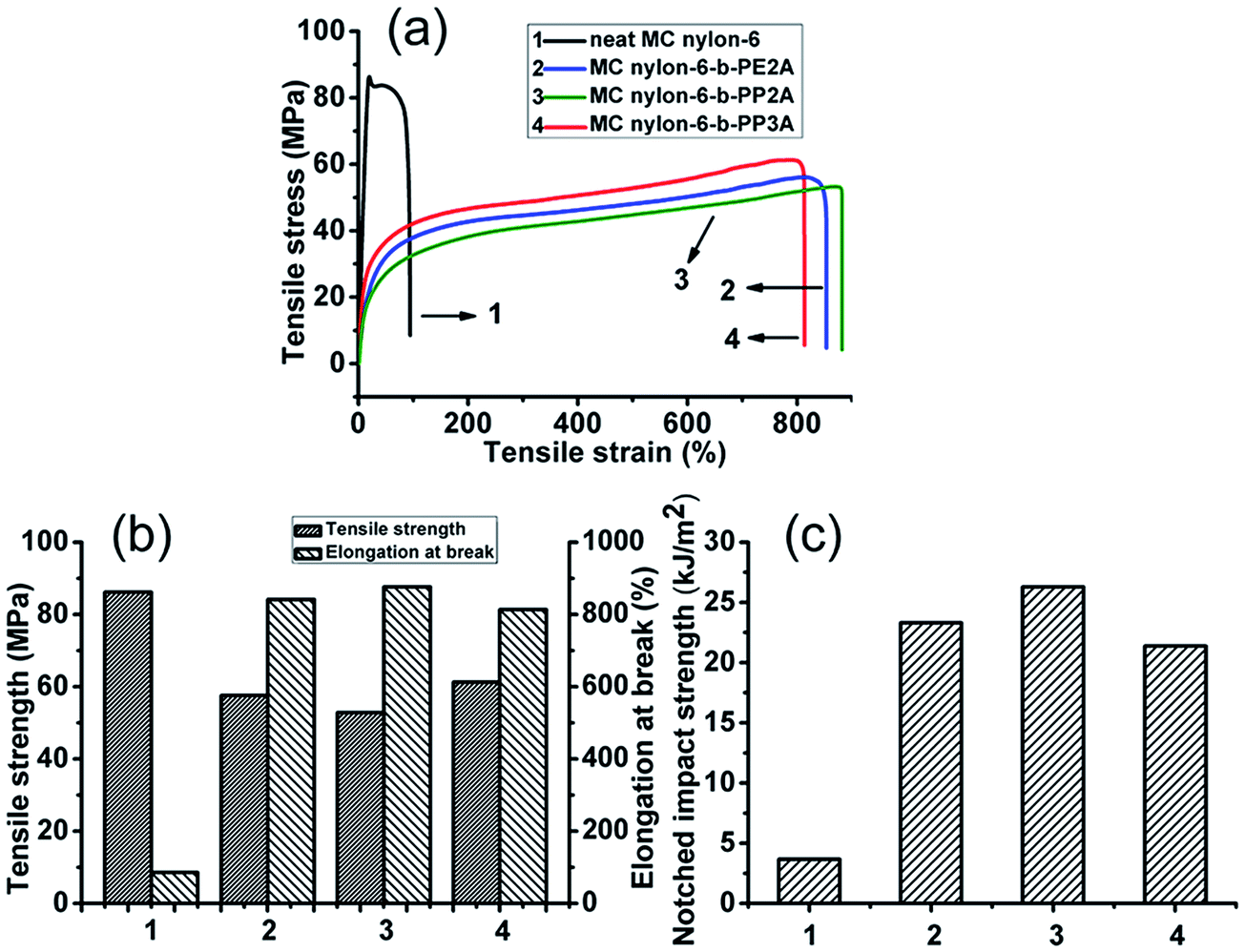
3.4.1 Static mechanical properties. Fig. 6(a) shows the tensile stress–strain curves of neat MC nylon-6 and the MC nylon-6-b-polyether amine copolymers. It can be seen that the stress–strain curves of the neat MC nylon-6 presented characteristics of brittle fracture with a typical yield point and low elongation at break. However, with the introduction of polyether amine, the area under the stress–strain curves of the copolymers increased dramatically, indicating a significantly increasing toughness of the copolymers. The stress–strain curves of all copolymers showed an untypical yield point and presented obvious strain hardening behavior after a long elastic deformation stress plateau.
 |
| | Fig. 6 Mechanical properties of the MC nylon-6-b-polyether amine copolymers. | |
Fig. 6(b and c) show the mechanical properties of all samples. It can be seen that the elongation at break and notched impact strength of the copolymers were increased by 800% compared with neat MC nylon-6, indicating a notable toughening effect of the polyether amine on the nylon-6 matrix, while the tensile strength of the copolymers is still maintained at a high level. Among the copolymers, the tensile strength of MC nylon-6-b-PP3A was the highest and the elongation at break and notched impact strength were lowest due to its high crystallinity and significant physical entanglement of much more branched molecular chains. The relatively low degree of molecular regularity of MC nylon-6-b-PP2A resulted in a low tensile strength and high notched impact strength.
3.4.2 Dynamic mechanical analysis. The numerical DMA data of neat MC nylon-6 and the MC nylon-6-b-polyether amine copolymers are listed in Table 5. It can be seen that, compared with neat MC nylon-6, the values of the loss factor of the copolymers were improved significantly and the values of the storage modulus decreased dramatically, indicating that the movement of the copolymer’s molecular chains was easy and the internal friction increased with the introduction of multi-branched molecular chains. The tanδ curve of neat MC nylon-6 showed two relaxation peaks at −62 °C and 50 °C corresponding to the β relaxation and α relaxation, respectively. The α relaxation arose from the chain segmental motion of molecules in the amorphous region and corresponded to the glass transition temperature (Tg). The high glass transition temperature of neat MC nylon-6 indicates rigid molecular chains. In the case of the MC nylon-6-b-polyether amine copolymers, the α relaxation peak dramatically shifted to a low temperature while the change in the β relaxation was not obvious. Moreover, the MC nylon-6-PP3A presented the highest storage module and glass transition temperature, and lowest loss factor among the copolymers, which may be benefits of its high crystallinity and high physical entanglement of molecular chains.
Table 5 Numerical DMA data for the MC nylon-6-b-polyether amine copolymer
| Sample |
Storage modulus (MPa) |
α peak (°C) |
β peak (°C) |
Loss factor tanδ |
| Neat MC nylon-6 |
5090 |
50.2 |
−62.6 |
0.137 |
| MC nylon-6-b-PE2A |
3446 |
7.3 |
−64.6 |
0.155 |
| MC nylon-6-b-PP2A |
3104 |
7.4 |
−63.1 |
0.156 |
| MC nylon-6-b-PP3A |
3930 |
22.5 |
−61.3 |
0.150 |
3.5 Toughening mechanism of the MC nylon-6-b-polyether amine copolymers
Fig. 7(a) shows a cryogenically fractured surface of a MC nylon-6-b-polyether amine copolymer. It can be seen that it was difficult to distinguish the interface between the two phases, which suggests that the MC nylon-6 phase and polyether amine phase were compatible. The polyether amine was covalently connected with nylon-6 molecules. Unlike the simple blending with a high molecular weight soft component, the phase separation in excess of a micrometer scale between the two components had difficulty occurring due to the copolymerization method and low molecular weight of the polyether amine.
 |
| | Fig. 7 SEM images of a cryogenically fractured surface (a: magnification ×10000) and impact fractured surfaces of MC nylon-6-b-polyether amine copolymers (b–e: magnification ×500; b′–e′: magnification ×5000). | |
The fracture surface morphology of the samples was also studied using SEM. As seen in Fig. 7(b–e) at low magnification (×500), the fracture surface of neat MC nylon-6 was relatively smooth and showed a typical brittle fracture, while that of the copolymers was rough and accompanied by a large number of major deformations from absorbing the impact energy, displaying characteristics of tough fracture. In addition, an obvious hairy structure was observed on the fracture surfaces of all copolymers. To further observe the fracture surfaces, images at a high magnification (×5000) were taken, as seen in Fig. 7(b′–e′), where it could be seen that the hairy structure on the fracture surface was acicular fibers of the nylon-6 matrix which were formed in a way similar to wire-drawing of the matrix under shear stress. The hairy structure of MC nylon-6-b-PP2A was the thickest and longest among the copolymers, which was coincident with the impact strength result.
After undergoing shear stress, many acicular fibers formed on the impact fracture surface of the samples and the fibers were connected to the matrix resin. The front part of the acicular fibers showed a whitening phenomenon while the bottom part adjacent to the matrix resin was relatively dark. The toughening mechanism of this copolymer system can be deduced to be a multi-layer crack extension mechanism. As shown in Fig. 8, certain stress defects in the nylon-6 matrix induced the formation of multi-layer micro-cracks as the stress concentration points, which quickly developed into a deep micro-crack vacuum layer field with a face to face adhesive sheet film. Under shear stress, the molecular chains of the adhesive sheet film oriented intensively along the orientation of the stress and then developed into the hairy structures with oriented-long fibers, which broke when the impact energy was higher than the surface energy and the molecular orientation energy. The whitening phenomenon in the front part of the fibers confirmed the intensive orientation of the fibers under shear stress. Therefore, different from the usual craze–shear band mechanism, the super toughening break of the MC nylon-6-b-polyether amine copolymers was based on the energy consumption of the extension of quantity of adhesive sheet films and the orientation of the molecular chains in the matrix fibers, not only the result of the energy consumption of the craze–shear band.
 |
| | Fig. 8 Toughening mechanism of the MC nylon-6-b-polyether amine copolymers. | |
4. Conclusions
MC nylon-6-b-polyether amine copolymers with different molecular structures were prepared via in situ polymerization. The effect of the molecular structure of the polyether amine on the synthesis and properties of the copolymers was studied. The result showed that the introduction of polyether amine dramatically delayed the polymerization process of caprolactam due to the steric effect of multi-armed long-chain structures, and the value of E and A of the reaction increased for the copolymers. The hydrogen bond and inter-molecular forces of the copolymers were weakened by the introduction of multi-branched molecular chains. The crystallization process of the copolymers produced the α-form mainly and gradually formed a small amount of γ-form simultaneously. The crystallization ability decreased for the copolymers, among which MC nylon-6-b-PP3A showed the highest melting and crystallization temperatures and crystallinity with relatively large crystal grain size due to the crystallizability of the extra branched-chain. A pseudo-plastic fluid characteristic of flow behavior was observed for MC nylon-6. The G and νe of all copolymers were higher than that of neat MC nylon-6 due to the severe entanglement through the formation of physical networks; MC nylon-6-b-PP3A presented the highest. The loss factor of the copolymers increased, whereas the storage modulus and Tg decreased. The area under the stress–strain curves of the copolymers increased dramatically, indicating significantly increasing toughness of the copolymers. A hairy fibrous structure was observed on the fracture surface of the copolymers, displaying characteristics of tough fracture. The toughening mechanism of the copolymers was deduced as a multi-layer crack extension mechanism.
References
- S. Y. Zeng, X. Lin and C. Liu, Production of MC nylon rod using horizontal centrifugal casting technique, Eng. Plast. Appl., 2011, 39(10), 52–54 CAS.
- F. J. Zhou, L. Q. Zhang and Y. M. Wang, Synthesis of MC Nylon, Synthetic Technology and Application, 2012, 27(2), 1–6 Search PubMed.
- X. Lin, P. N. Zhang, Z. L. Yin and Q. Y. Chen, Study on preparation and properties of La2O3/MC nylon nanocomposites, J. Rare Earths, 2005, 23(6), 680–684 Search PubMed.
- Y. M. Zhang, X. Y. Wu, W. Hong and W. Zhang, Toughening properties of MC nylon with UHMWPE particle, Plastics, 2009, 38(1), 68–69 CAS.
- X. H. Wang, Q. Zhang and X. B. Zhang, A study of the property characterization of MC copolymerized nylon elastomer, China Plast. Ind., 1996, 24(6), 55–58 CAS.
- K. K. Jin, K. Y. Young, Y. B. Sook and Y. K. Jong, J. Appl. Polym. Sci., 1995, 57, 1347–1357 CrossRef.
- B. B. Wang and G. S. Hu, Preparation and characterization of nylon 6 11 copolymer, Mater. Lett., 2006, 60(21), 2715–2717 CrossRef CAS PubMed.
- C. S. Liu and Q. Wang, New method on preparing of nylon 6 micro-powder and PA 6/PP/wollastonite composite, J. Wuhan Inst. Chem. Technol., 2001, 23(3), 35–39 Search PubMed.
- S. Xu, X. W. Zhao and L. Ye, Preparation and Properties of Monomer Casting Nylon-6/PEO Blend Prepared via In-situ Polymerization, Polym. Eng. Sci., 2013, 53(9), 1809–1822 CAS.
- B. L. Pan, S. P. Zhang, W. Z. Li, J. Z. Zhao, J. L. Liu, Y. Q. Zhang and Y. Z. Zhang, Tribological and mechanical investigation of MC nylon reinforced by modified graphene oxide, Wear, 2012, 294(30), 395–401 CrossRef PubMed.
- G. Rusu, K. Ueda, E. Rusu and M. Rusu, Polyamides from lactams by centrifugal molding via anionic ring-opening polymerization, Polymer, 2001, 42(13), 5669–5678 CrossRef CAS.
- C. H. Lu, P. Y. Yeh and W. T. Hsu, Non-isothermal reaction kinetics of lithium cobalt oxide, J. Alloys Compd., 2009, 476(1), 749–754 CrossRef CAS PubMed.
- J. M. Schultz, B. S. Hsiao and J. M. Samon, Structural development during the early stages of polymer melt spinning by in-situ synchrotron X-ray techniques, Polymer, 2000, 41(25), 8887–8895 CrossRef CAS.
- J. C. Ho and K. H. Wei, Induced γ→α Crystal Transformation in Blends of Polyamide 6 and Liquid Crystalline Copolyester, Macromolecules, 2000, 33(14), 5181–5186 CrossRef CAS.
- J. D. Ferry, Viscoelastic Properties of Polymers, Wiley, New York, 1980 Search PubMed.
- M. Doi and S. F. Edwards, Dynamics of concentrated polymer systems, part 3 the constitutive equation, J. Chem. Soc., Faraday Trans., 1978, 74, 1818–1832 RSC.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.