
Water steam effect during high CO2 chemisorption in lithium cuprate (Li2CuO2) at moderate temperatures: experimental and theoretical evidence
Abstract
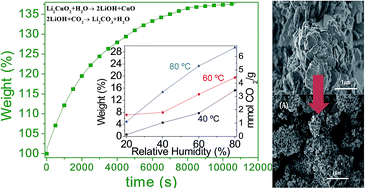
Li2CuO2 was evaluated as a CO2 captor at moderate temperatures, using water vapor in the gas flow. Different water vapor sorption experiments were performed using N2 or CO2 as carrier gases. If N2 was used as carrier gas, it was evidenced that Li2CuO2 is able to trap water physically and chemically, producing in the second case Li–OH superficial species. Moreover, when CO2 was used as carrier gas, Li2CuO2 continued trapping water, as in the previous case, but in this case CO2 was mainly trapped, forming Li2CO3 and CuO phases. Additionally, the microstructure changes importantly when CO2 and H2O are chemically trapped in Li2CuO2. Li2CO3 and CuO seemed to segregate changing the morphology and the specific surface area. The Li2CuO2 sample was able to capture up to 6.7 mmoles of CO2 per gram of ceramic at 80 °C, a considerably high CO2 amount. Furthermore, all these experiments were theoretically supported by different thermodynamic calculations. Experimental and theoretical results show that H2O acts as a catalytic intermediate, diminishing the activation energy of the whole CO2 chemisorption process. Therefore, the presence of water vapor strongly favored the CO2 chemisorption on Li2CuO2 at moderate temperatures (30–80 °C).
 Please wait while we load your content...
Please wait while we load your content...

