
Comparative study on confinement effects of graphene and graphene oxide on structure and dynamics of water
Abstract
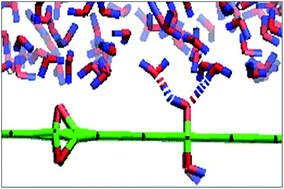
In this work, we compared the structural and dynamical properties of water confined between two graphene oxide (GO) sheets with water confined between two graphene sheets through molecular dynamics simulations. Our results showed how the structure and dynamics of the water near the GO surfaces changes under confinement conditions. Different orientations of the water molecules to the GO sheets confirm the heterogeneous nature of the confined water. The density distribution of water near the GO sheets is different from the graphene surfaces due to the presence of hydrophilic functional groups of the GO sheets. Also, the hydrogen bonds between the water molecules are disturbed due to the presence of these groups on the GO surfaces. The results showed that as water molecules in areas which are close to the GO surfaces are isolated, and due to hydrogen bond formation between water molecules and the substituents of the GO, the water molecules have fewer hydrogen bonds with other water molecules around. In the middle region between two GO sheets, it has been seen that each water molecule is surrounded by more water molecules, and there are many more hydrogen bonds, so it is possible to consider a network structure of water in this region. There is a good quantitative agreement between experiments and the above mentioned results. These results were confirmed by the calculated coordination number, radial distribution functions and mean square displacements.
 Please wait while we load your content...
Please wait while we load your content...

