
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, biological evaluation and molecular modeling of new analogs of the anti-cancer agent 2-methoxyestradiol: potent inhibitors of angiogenesis†
Eirik Johansson
Solum
a,
Jing-Jy
Cheng
*bc,
Ingebrigt
Sylte
d,
Anders
Vik
a and
Trond Vidar
Hansen
*a
aDepartment of Pharmaceutical Chemistry, School of Pharmacy, University of Oslo, PO Box 1068 Blindern, N-0316 Oslo, Norway. E-mail: e.j.solum@farmasi.uio.no; anders.vik@farmasi.uio.no; t.v.hansen@farmasi.uio.no; Fax: +47 22855947; Tel: +47 22857450
bNational Research Institute of Chinese Medicine, 155-1 Li-Nung Street, Section 2, Shih-Pai, Taipei, Taiwan. E-mail: verona@nricm.edu.tw; Fax: +886-2-2825-0743; Tel: +886-2-2820-1999-3671
cInstitute of Biophotonics, National Yang-Ming University, Taipei 112, Taiwan
dDepartment of Medical Biology, Faculty of Health Sciences, UiT – The Arctic University of Norway, 9037 Tromsø, Norway. E-mail: ingebrigt.sylte@uit.no
First published on 27th March 2015
Abstract
The synthesis, cytotoxicity, inhibition of tubulin polymerization and anti-angiogenic effects of 10 analogs of 2-methoxyestradiol are reported. These efforts revealed that the analog with a 4-pyridine ring in the 17-position, in combination with 2-ethyl- and 3-sulfamate substituents on the steroid A-ring, is the most interesting anti-cancer agent. This compound showed potent inhibitory effects against angiogenesis (IC50 = 0.1 ± 0.02 μM) and selective cytotoxic effects towards the CEM, H460 and HT-29 cancer cell lines, with no cytotoxicity observed against the healthy VERO cell line. The most interesting analog also displayed inhibition of tubulin polymerization (IC50 = 4.3 μM) almost as potent as 2-methoxyestradiol (IC50 = 3.5 μM). Molecular modeling experiments showed that this analog interacts within the colchicine-binding site of β-tubulin via multiple bonding with several amino acids. These observations provide support that the cytotoxic and anti-angiogenic effects observed for this novel analog are, at least in part, mediated by binding to tubulin.
Introduction
Several steroids, exemplified by compounds 1–6 in Fig. 1, display anti-cancer effects and some have entered the drug market. The endogenous steroid 2-methoxyestradiol (2-ME, 1) exhibits anti-vascular effects1 and anti-angiogenic activities.2 In 1989, Seegers et al. reported that high micro-molar concentrations of 1 affected dividing cancer cells.3 Five years later, D'Amato et al. reported that 1 was a tubulin polymerization inhibitor and a competitive inhibitor of colchicine.4 A number of biological studies followed which showed that the steroid 1 possesses many interesting anti-cancer effects without any undesirable estrogen activity.5,6 2-ME (1) has entered several clinical trial development programs,7 and some structural-activity relationship (SAR) studies have been conducted with 1 as the lead compound.8 ENMD-1198 (2) is one example that emerged from these efforts.9 Another anti-cancer steroid is abiraterone (3) that is used, as its acetate prodrug, in combination with prednisone (4) against metastatic castration-resistant prostate cancer. Abiraterone (3) is marketed under the trade name Zytiga™,10 see Fig. 1.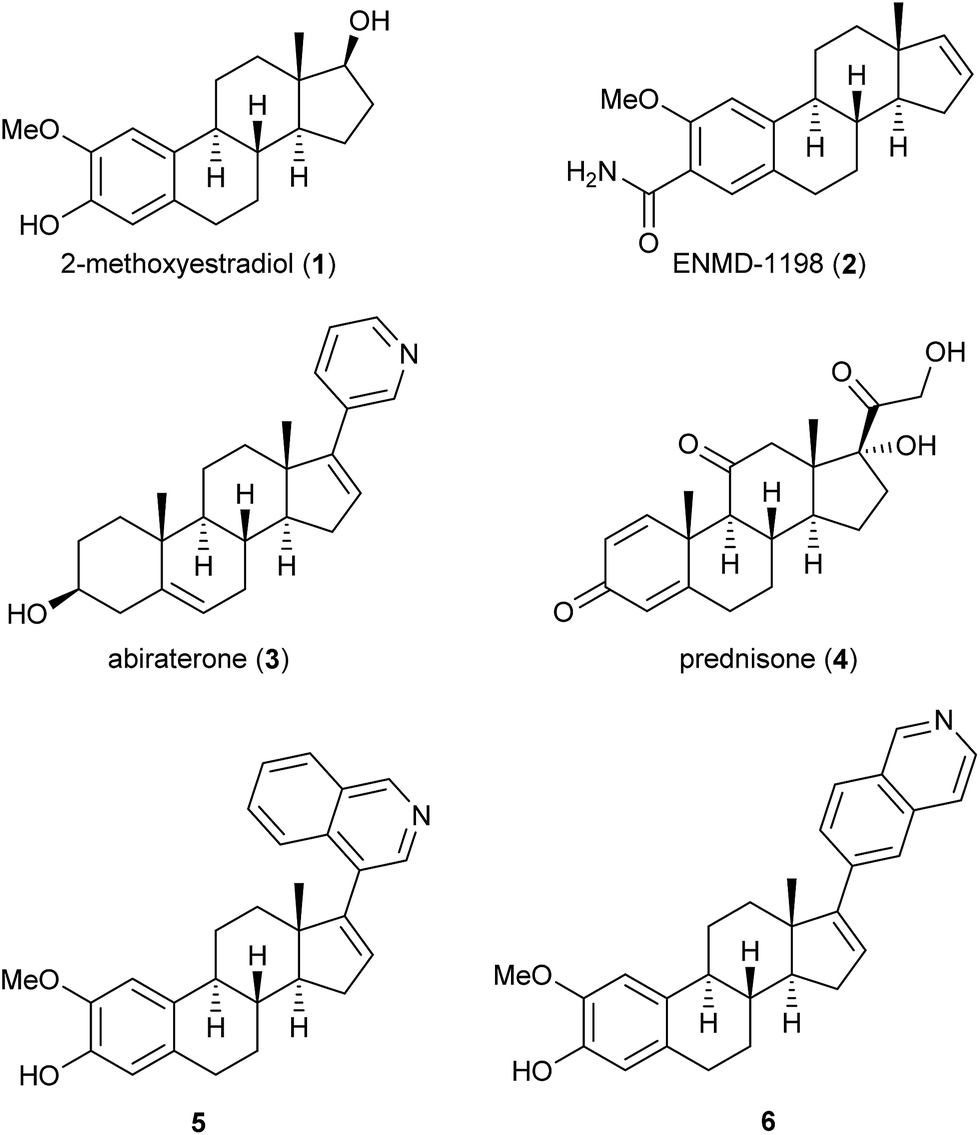 | ||
| Fig. 1 Examples of steroids with anti-cancer effects. | ||
Recently we reported that the two compounds 5 and 6, see Fig. 1, with a 4- or a 6-substituted isoquinoline ring in the 17-position of the steroid skeleton of 2-ME (1), respectively, showed inhibition of tubulin polymerization and anti-angiogenic effects in the low micro-molar range.11 It has been reported that substituting the methoxy group with an ethyl group in the 2-position of 1 has resulted in new analogs with interesting anti-cancer effects.12 Based on our previous findings, we wanted to conduct a SAR-study substituting the 2-methoxy group with an ethyl group, as well as introducing an aryl moiety in the 17-position of 1. Previously, it has also been reported that replacing the phenol in the A-ring of 2-ME (1) with a sulfamate group has resulted in enhanced cytotoxicity.13 Hence, we wanted to include also this substituent in our studies. Overall, this resulted in the design of the novel steroids 7a–7e and 8a–8e. The synthetic work, molecular modeling studies and the biological evaluation of these novel 2-ME (1) analogs are presented herein.
Results and discussions
Chemistry
The synthesis of compounds 7a–7e and 8a–8e commenced with the ortho-formylation reaction14 with estradiol 9 as the substrate, in the presence of a mixture of para-formaldehyde, MgCl2 and Et3N in refluxing THF. As previously reported,15 the regioisomeric ratio was observed to be 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 in favor of the desired salicylaldehyde 10. Regioisomeric pure product was obtained in 81% yield after chromatography. Then a Wittig-reaction between 10 and the yield of methyltriphenylphosphonium bromide, the latter obtained after reaction with sodium tert-butoxide, afforded the styrene 11. Reduction of the double bond in 11 with hydrogen in the presence of palladium on carbon gave the desired 2-ethyl substituted estradiol 12 in 61% yield over the three steps (Scheme 1). Further modification of the 17-position was achieved in a three-step protocol. Oxidation of 12, followed by TBS-protection of the phenol in 13, yielded the ketone 14. Compound 14 was converted, as previously reported,16 to the enol triflate 15. The triflate 15 was reacted in a Suzuki–Miyaura cross-coupling reaction with the enumerated boronic acids (Scheme 1), affording the desired products 16a–16e in 73–84% yields. Finally, deprotection of the TBS-group using an excess of tetra-n-butyl ammonium fluoride followed by purification by column chromatography yielded the desired 2-ethyl estrone analogs 7a–7e as stereoisomeric pure products. The introduction of the sulfamate in the 3-position of 1 was achieved by reacting the phenol with sulfamoyl chloride in the presence of 2,6-di-tert-butyl-4-methylpyridine (DBMP) in dichloromethane as solvent. The products 8a–8e were obtained in 69–76% yields.
:1 in favor of the desired salicylaldehyde 10. Regioisomeric pure product was obtained in 81% yield after chromatography. Then a Wittig-reaction between 10 and the yield of methyltriphenylphosphonium bromide, the latter obtained after reaction with sodium tert-butoxide, afforded the styrene 11. Reduction of the double bond in 11 with hydrogen in the presence of palladium on carbon gave the desired 2-ethyl substituted estradiol 12 in 61% yield over the three steps (Scheme 1). Further modification of the 17-position was achieved in a three-step protocol. Oxidation of 12, followed by TBS-protection of the phenol in 13, yielded the ketone 14. Compound 14 was converted, as previously reported,16 to the enol triflate 15. The triflate 15 was reacted in a Suzuki–Miyaura cross-coupling reaction with the enumerated boronic acids (Scheme 1), affording the desired products 16a–16e in 73–84% yields. Finally, deprotection of the TBS-group using an excess of tetra-n-butyl ammonium fluoride followed by purification by column chromatography yielded the desired 2-ethyl estrone analogs 7a–7e as stereoisomeric pure products. The introduction of the sulfamate in the 3-position of 1 was achieved by reacting the phenol with sulfamoyl chloride in the presence of 2,6-di-tert-butyl-4-methylpyridine (DBMP) in dichloromethane as solvent. The products 8a–8e were obtained in 69–76% yields.
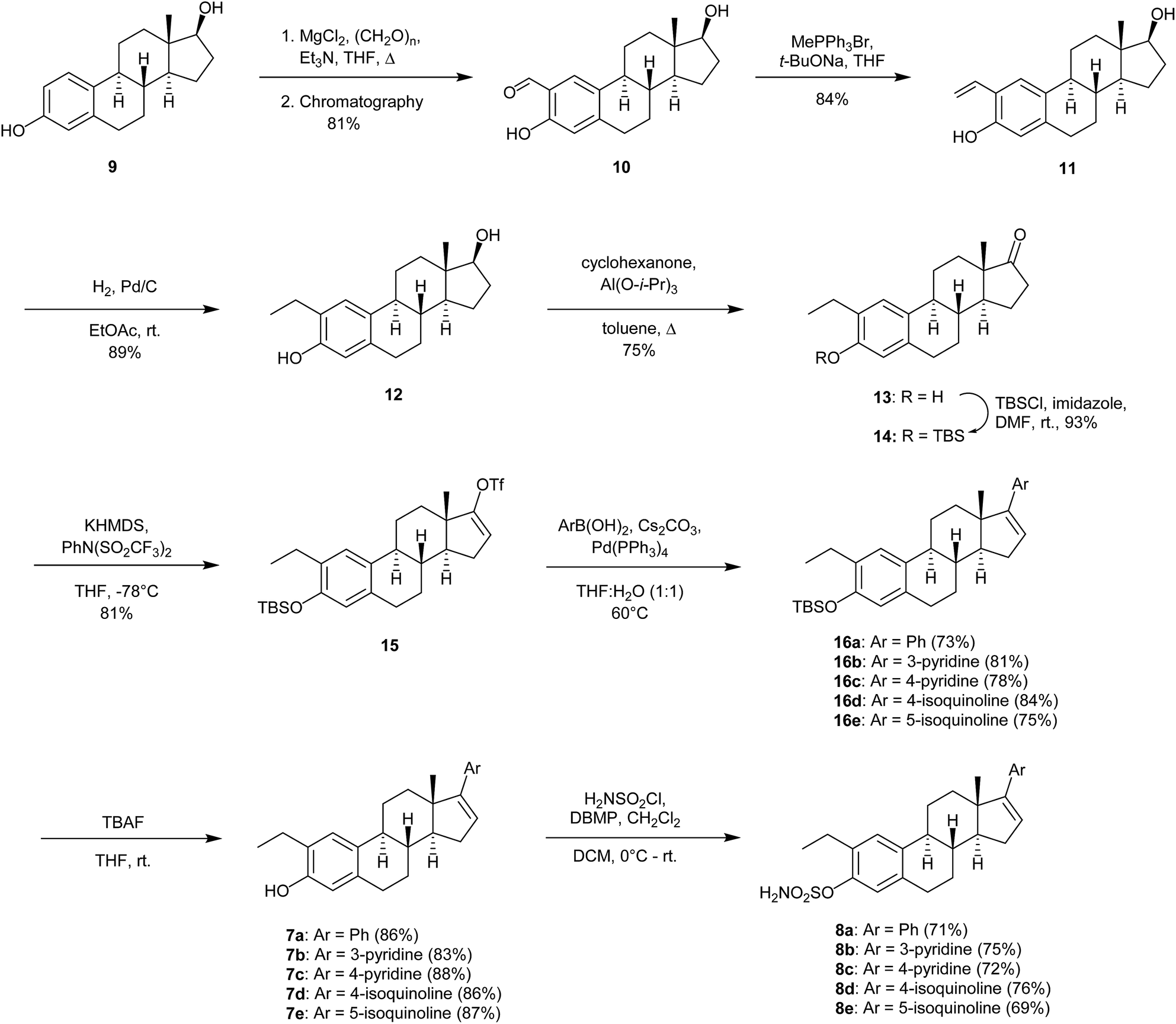 | ||
| Scheme 1 Synthesis of estrogen analogs 7a–7e and 8a–8e. | ||
Biological evaluations
| Compound | CEM cell assay IC50a (μM) | H460 cell assay IC50a (μM) | HT-29 cell assay IC50a (μM) | VERO cell assay IC50a (μM) | Anti-angiogenesis IC50a (μM) | Tubulin polymerization inhibitionb (%) | Tubulin polymerization inhibition IC50c (μM) |
|---|---|---|---|---|---|---|---|
| a Results of three experiments performed as triplicates. b Determined at 10 μM. c Results of two experiments performed as triplicates. d n.d = not determined. | |||||||
| Colchicine | n.d. | n.d. | n.d. | n.d. | n.d. | 100 | n.d.d |
| Paclitaxel | n.d. | n.d. | n.d. | n.d. | n.d. | 0 | n.d. |
| 7a | 12.9 ± 2.1 | 16.9 ± 3.1 | 19.3 ± 2.3 | 29.8 ± 2.6 | >10 | 41 | n.d. |
| 7b | 66.9 ± 6.1 | >256 | >256 | >256 | >10 | 51 | n.d. |
| 7c | >256 | >256 | >256 | >256 | >10 | 38 | n.d. |
| 7d | >256 | >256 | >256 | >256 | >10 | 54 | n.d. |
| 7e | 160.9 ± 23.5 | >256 | >256 | >256 | >10 | 58 | n.d. |
| 8a | 5.4 ± 0.7 | 9.3 ± 2.3 | 7.9 ± 1.1 | 9.3 ± 1.7 | >10 | 109 | 8.1 |
| 8b | 86.5 ± 9.8 | >256 | 49.7 ± 5.9 | >256 | 0.2 ± 0.03 | 158 | 6.1 |
| 8c | 8.0 ± 1.4 | 110.3 ± 13.6 | 28.3 ± 3.7 | >256 | 0.1 ± 0.02 | 109 | 4.3 |
| 8d | 46.2 ± 5.4 | >256 | >256 | >256 | 0.7 ± 0.04 | 36 | 7.7 |
| 8e | 67.0 ± 9.5 | >256 | 160.9 ± 18.5 | >256 | >10 | 56 | n.d. |
| 2-ME (1) | 84.9 ± 9.7 | 63.3 ± 7.1 | >256 | >256 | 3.2 ± 0.22 | 138 | 3.5 |
The two compounds 7a and 8a exhibited potent cytotoxic effects against all of the cancer cell lines. Unfortunately, these compounds also inhibited the growth of the VERO cell line. The phenyl ring attached at the 17-position of the steroid skeleton is apparently detrimental for any selective inhibition towards cancer cell growth. Among the other analogs tested, several of the compounds proved to be active, especially towards the human CEM leukemia cell line. Noteworthy, all of the compounds except 2-ME (1) exhibited lower activity towards the lung cancer cell line H460 and the colon cancer cell line HT-29. Compound 8c showed good activity towards the CEM leukemia cell line (IC50 = 8.0 ± 1.4 μM). To our delight, this compound did not exhibit any activity towards the VERO cell line. Analogs 8a–8e exhibited better cytotoxic properties than 7a–7e. Among these analogs, the only active analogs were compounds 7b and 7e that showed cytotoxic effects in the CEM cell line. The introduction of the 2-ethyl and the sulfamate substituents gave better selectivity as well as cytotoxicity against the human CEM leukemia cell line.
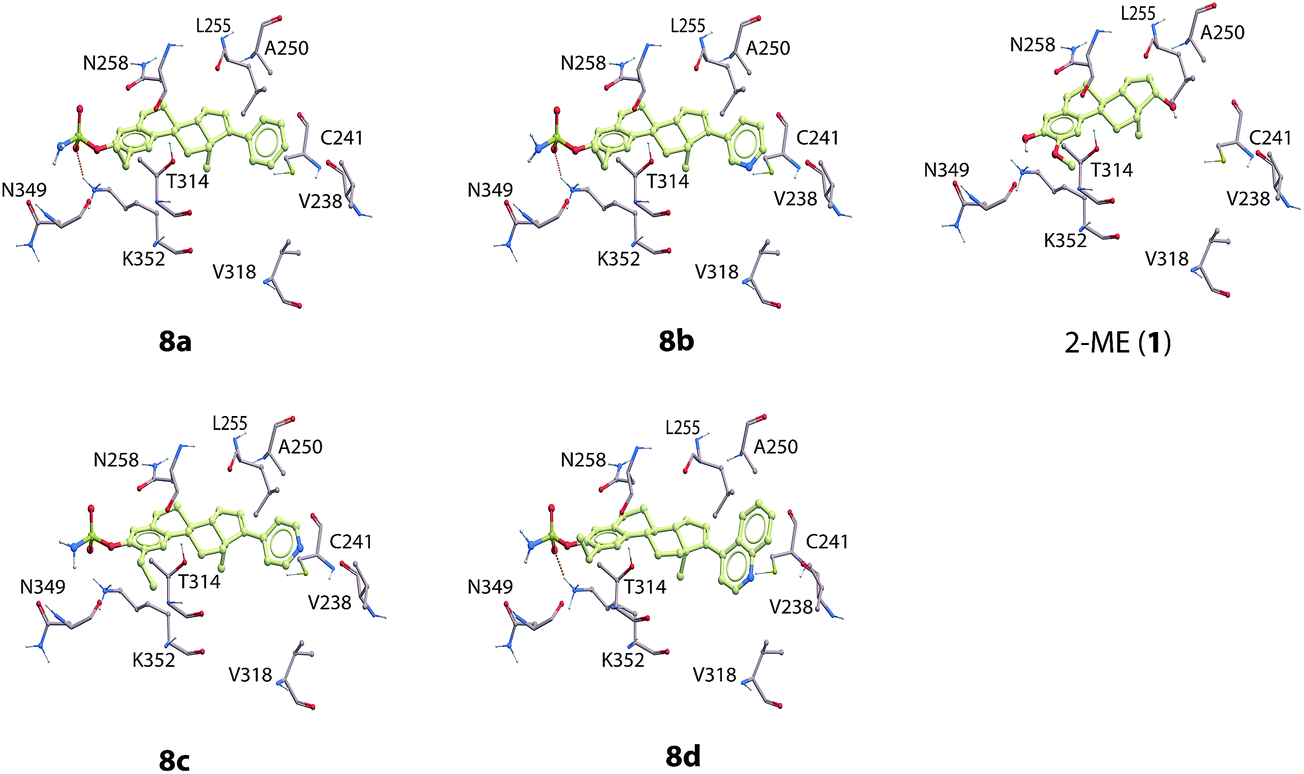 | ||
| Fig. 2 The docked compounds (8a–8d and 2-ME (1)) in the colchicine binding site of β-tubulin. The most important amino acids for ligand binding are included in the figure. Color coding of atoms in the compounds and the amino acids: red; oxygen, blue; nitrogen, yellow: carbon, dark-yellow: sulfur. Ligand hydrogen atoms are not displayed. | ||
The molecular modeling studies of the novel analogs showed that the sulfamate group present in compounds 8a–8d interacts within a pocket consisting Asn349 (backbone oxygen), Asn258 (side chain oxygen) and Lys352 (terminal side chain hydrogens). Hydrogen bonding interactions were formed between the side chain of Lys352 and the sulfamate group (Fig. 2) and all compounds 8a–8d. The 2-ethyl group of 8a–8d form hydrophobic interactions with Thr314, Val315, Asn350 and the side chain of Lys352. The phenyl group of 8a, the 3-pyridine group of 8b, the 4-pyridine group of 8c, and the 4-isoquinoline group of 8d all interacted in a hydrophobic pocket consisting of Val238, Cys241, Leu248, Ala250, Leu255, Ala316, Val318, Ala354, and Ile378.
Discussions
The introduction of an ethyl group in the 2-position of 2-ME (1) has previously been reported to afford potent inhibitors against angiogenesis.12,23 This knowledge, together with the introduction of new analogs substituted in the 17-position, gave several potent cytotoxic agents that also inhibited tubulin depolymerization. Moreover, introducing the sulfamate group in the 3-position enhanced both the cytotoxicity and the tubulin inhibition, but also resulted in better inhibition of angiogenesis, as seen for compounds 8a–8d. Overall, compound 7a displayed the most potent cytotoxic effects. However, towards the development of new anti-cancer agents, the most promising candidate is compound 8c. This compound showed cytotoxic effects in all three cancer cell lines, but no such effects against the VERO cells. In addition, this compound also exhibited very potent anti-angiogenic activities in the nanomolar range. Corey and co-workers have reported that the position of the nitrogen atom in the heterocyclic substituent at the C-17 position in some steroids is essential for potent anti-angiogenesis activity.24 Moreover, It has been reported that sterols interacts with several proteins and biological targets.25 The compounds reported herein may exhibit their mode of actions by interacting with multiple biological targets. The anti-angiogenic activities displayed by the new analogs reported herein are also dependent on the substitution pattern. Compound 8c also displayed inhibition of tubulin polymerization (IC50 = 4.3 μM) in the same range as for 2-ME (1), IC50 = 3.5 μM. Compounds 8a, 8b and 8d showed slightly less effects towards inhibition of tubulin than both 1 and 8c.The molecular docking showed that the virtual ligand screening (VLS) scoring function values for of compounds 8a–8d with tubulin were all in the same range as the scoring of 2-ME. The new analogs 8a–8d exhibited similar binding mode in the colchicine binding pocket as the lead compound 2-ME (1) (Fig. 2). These observations were also reflected in the comparable IC50-values obtained from the tubulin inhibition studies, see Table 1. However, compound 8a had a slightly better scoring in tubulin than 8a–8d indicating that changing the phenyl group of compound 8a into a 3- or a 4-substituted pyridine ring, as for 8b and 8c, respectively, or into a 4-isoquinoline group as for 8d, decreased the binding affinity to β-tubulin compared to 8a. These parts of the compounds interact in a hydrophobic region of β-tubulin and the phenyl group of 8a seems to obtain more favorable interactions than the nitrogen containing ring systems (Fig. 2).
Conclusion
In total, 10 new analogs of 2-ME (1) have been prepared using the ortho-formylation and the Suzuki–Miyaura reactions. All analogs were evaluated for their cytotoxic effects, as well as their anti-angiogenic activity and inhibition of tubulin polymerization. Compound 8c exhibited more potent cytotoxic and anti-angiogenic effects than 1. This compound has a sulfamate group in the 3-position of ring A in the steroid skeleton. Such sulfamates of estrogens have been reported to be multi-targeted anti-cancer agents,22 and the biological effects of the new analogs reported herein may also arise via such mechanisms. Such types of anti-cancer agents are of current interest towards the potential development of remedies against cancer, including leukemia.26 The results presented herein provide new information on such new lead compounds.Experimental
General methods
All reagents and solvents were used as purchased without further purification unless stated otherwise. Melting points are uncorrected. Analytical TLC was performed using silica gel 60 F254 aluminum plates (Merck). Flash column chromatography was performed on silica gel 60 (40–63 μm) produced by Merck. NMR spectra were recorded on a Bruker Avance DPX-300 MHz, DPX-400 MHz or DPX-600 MHz spectrometer for 1H NMR, and 75 MHz, 101 MHz or 151 MHz for 13C NMR. Coupling constants (J) are reported in hertz, and chemical shifts are reported in parts per million relative to CDCl3 (7.26 ppm for 1H and 77.0 ppm for 13C). Mass spectra were recorded at 70 eV with Fison's VG Pro spectrometer. High-resolution mass spectra were performed with a VG Prospec mass spectrometer and with a Micromass Q-TOF-2™. The HPLC analyses were performed on an Agilent Technologies 1200 Series instrument with an Eclipse XDB-C18 (5 mm 4.6 × 150 mm) column. Optical rotations were measured using a 1 ml cell with 1.0 dm path length on Perkin-Elmer 341 polarimeter in dedicated solvent. Protocols for the preparation, physical and spectral data of the intermediates 10–16 and products 7a–7e and 8a–8e are presented in the ESI.†Cancer cell growth inhibition
To assess cell viability, the AlamarBlue® (AB) assay (dye purchased from Biosource International, Nivelles, Belgium) was used as previously described.11,17 This involved aspirating medium at the end of each treatment period and adding 100 μl of fresh medium containing 10% v/v AB to control and treated wells. Plates were incubated at 37 °C for six hours prior to measuring the absorbance at 540 nm and at 595 nm wavelengths using a spectrophotometric plate reader (DYNEX Technologies, USA). Experimental data were normalized to control values.Inhibition of tubulin polymerization
The method applied was that described by Lawrence and coworkers.18 The assay was performed using a commercial kit (Cytoskeleton Inc., Denver, USA). Briefly, samples were prepared directly in a 96-well microtiter plate that was pre-incubated at 4 °C in the fridge for 30 min and contained Mes buffer [128 μl (0.1 M Mes, 1 mM EGTA, 0.5 mM MgCl2, distilled water, pH 6.6)], GTP (20 μl, 5 mM in Mes buffer), tubulin (50 μl, 11 mg ml−1 in Mes buffer) and the test compound (20 μl in DMSO). The tubulin and samples of test compounds were immediately placed in a 96-well plate reader, alongside the blank samples containing Mes buffer (198 μl) and the analogs (10 μl, same concentration). The absorbance (λ 340 nm) was recorded at 25 °C temperature for a period of 60 min. The polymerization curve was made as OD of each sample (Y axis) vs. recording time (X axis). The AUC (area under curve) between zero to 30 minutes was obtained to present the polymerization degree using Sigmaplot software. After AUC was obtained, the average AUC was calculated (see ESI†) to get the inhibition percentage. Colchicine was set as 100% inhibition and paclitaxel as 0%. The IC50-value was calculated after obtaining the curve equation of inhibition % (Y axis) and concentration (X axis) using Excel.Inhibition of angiogenesis
The method applied was essential that described previously.17 Endothelial cell tube formation assay was modified from a method previously described.11 Matrigel (12.5 mg ml−1) was thawed at 4 °C, and 50 μl was quickly added to each well of a 96-well plate and allowed to solidify for 10 min at 37 °C. Once solid, the wells were incubated for 30 min with endothelial cell (30000 cells per well). After adhesion of the cells, the medium was removed and replaced by fresh medium supplemented with compounds with five different concentrations ranging from 10 μM to 0.001 μM and incubated at 37 °C for 18 h. The tubes of growth were visualized with an inverted ZEISS microscope at a magnification of 10. The tube formation areas were obtained using KURABO Angiogenesis Image Analysis Software. The length of the capillary network was quantified with a map scale calculator (KURABO Angiogenesis Image Analysis Software). Inhibition curve was obtained between areas and concentrations to get the IC50-value.
Molecular modelling
The Internal Coordinate Mechanics (ICM) program19 was used for docking of compounds 8a–8d and 2-ME (1) into the β-subunit of tubulin. The docking studies were performed with the tubulin structure from the X-ray complex of tubulin with colchicine (PDB id: 1SA0).20 Crystallographic water molecules, ions and co-crystallized inhibitors were removed from the X-ray complexes and hydrogen atoms were added and optimized using the ECEPP/3 force field of ICM. The compounds were built using ICM and optimized before docking. A grid map that included the amino acids within 5 Å of the co-crystallized inhibitors was calculated, and semi-flexible docking with flexible ligands was performed. Each docking was run in three parallels. The docking poses were scored using the Virtual ligand scoring (VLS) module of the ICM program.General procedure for the preparation of compounds 7a–e
The TBS protected steroids 16a–e (0.3–0.4 mmol, 1.0 equiv.) were placed in a dry round-bottomed flask under argon atmosphere, and dissolved in dry THF. Tert-butylamoniumfluoride (1 M in THF, 1.1 equiv.) was added drop vise. The reaction mixture was stirred at room temperature (16–18 h.). Upon completion the reaction the mixture was poured into saturated aqueous NaHCO3 (10 ml), and extracted with ethyl acetate (4 × 5 ml). The combined organic extracts were dried (MgSO4) and the solvent evaporated in vacuo. The residues were purified by chromatography (silica gel, 20–50% ethyl acetate in hexane) to give the pure products.General procedure for the synthesis of 8a–e
The estrogen (1 equiv.) and 2,6-di-tert-butyl-4-methylpyridine (3.0 equiv.) were added to a round bottomed flask and dissolved in dichloromethane under argon atmosphere. The mixture was cooled to 0 °C before sulfamoyl chloride (2.95 equiv.) was added. The reaction mixture was stirred at 0 °C for additional 30 min, and then at room temperature for 16–18 h. Sodium bicarbonate (saturated) was added and the mixture was extracted with EtOAc, dried over MgSO4 and evaporated. Flash column chromatography (50% EtOAc in hexane) afforded the products as colorless solids.Acknowledgements
The School of Pharmacy, University of Oslo, is gratefully acknowledged for a Ph.D. scholarship to E. J. S.References
- S. Bu, A. Blaukat, X. Fu, N. E. Heldin and M. Landstrom, FEBS Lett., 2002, 531, 141–151 CrossRef PubMed.
- S. Verenich and P. M. Gerk, Mol. Pharmaceutics, 2010, 7, 2030–2039 CrossRef PubMed.
- J. C. Seegers, M.-L. Aveling, C. H. van Aswegen, M. Cross, F. Koch and W. S. Joubert, J. Steroid Biochem., 1989, 32, 797–809 CrossRef PubMed.
- R. J. D'Amato, C. M. Lin, E. Flynn, J. Folkman and E. Hamel, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 3964–3968 CrossRef.
- T. Fotsis, Y. Zhang, M. S. Pepper, H. Adlercreutz, R. Montesano, P. P. Nawroth and L. Schweigerer, Nature, 1994, 268, 237–239 CrossRef PubMed.
- M. Sattler, L. R. Quinnan, Y. B. Pride, J. L. Gramlich, S. C. Chu, G. C Even, S. K. Kraeft, L. B. Chen and R. Salgia, Blood, 2003, 102, 289–296 CrossRef PubMed.
- (a) C. Sweeney, G. Liu, C. Yiannoutsos, J. Kolesar, D. Horvath, M. J. Staab, K. Fife, V. Armstrong, A. Treston, C. Sidor and G. Wilding, Clin. Cancer Res., 2005, 11, 6625–6633 CrossRef PubMed; (b) W. L. Dahut, N. J. Lakhani, J. L. Gulley, P. M. Arlen, E. C. Kohn, H. Kotz, D. McNally, A. Parr, D. Nguyen, S. X. Yang, S. M. Steinberg, J. Venitz, A. Sparreboom and W. D. Figg, Cancer Biol. Ther., 2006, 5, 22–27 CrossRef PubMed; (c) J. James, D. J. Murry, A. M. Treston, A. M. Storniolo, G. W. Sledge, C. Sidor and K. D. Miller, Invest. New Drugs, 2006, 25, 41–48 CrossRef PubMed; (d) S. V. Rajkumar, P. G. Richardson, M. Q. Lacy, A. Dispenzieri, P. R. Greipp, T. E. Witzig, R. Schlossman, C. F. Sidor, K. C. Anderson and M. A. Gertz, Clin. Cancer Res., 2007, 13, 6162–6167 CrossRef PubMed.
- J. F. Peyrat, J. D. Brion and M. Alami, Curr. Med. Chem., 2012, 19, 4142–4156 CrossRef PubMed.
- (a) C. Moser, S. A Lang, A. Mori, C. Hellerbrand, H. J. Schlitt, E. K. Geissler, W. E. Fogler and O. Stoeltzing, BMC Cancer, 2008, 8, 206–216 CrossRef PubMed; (b) E. Pasquier, S. Sinnappan, M. A. Munoz and M. Kavallaris, Mol. Cancer Ther., 2010, 9, 1408–1418 CrossRef PubMed.
- C. J. Ryan, M. R. Smith, J. S. de Bono, A. Molina, C. J. Logothetis, P. de Souza, K. Fizazi, P. Mainwaring, J. M Piulats, S. Ng, J. Carles, P. F. A. Mulders, E. Basch, E. J. Small, F. Saad, D. Schrijvers, H. Van Poppel, S. D. Mukherjee, H. Suttmann, W. R. Gerritsen, T. W. Flaig, D. J. George, E. Y. Yu, E. Efstathiou, A. Pantuck, E. Winquist, C. S. Higano, M.-E. Taplin, Y. Park, T. Kheoh, T. Griffin, H. I. Scher and D. E. Rathkopf, N. Engl. J. Med., 2013, 368, 138–148 CrossRef PubMed.
- E. J. Solum, J.-J. Cheng, I. B. Sørvik, R. E. Paulsen, A. Vik and T. V. Hansen, Eur. J. Med. Chem., 2014, 85, 391–398 CrossRef PubMed.
- S. P. Newman, M. P. Leese, A. Purohit, D. R. C. James, C. E. Rennie, B. V. L. Potter and M. J. Reed, Int. J. Cancer, 2004, 109, 533–540 CrossRef PubMed.
- A. B. Edsall, G. E. Agoston, A. M. Treston, S. M. Plum, R. H. McClanahan, T.-S. Lu, W. Song and M. Cushman, J. Med. Chem., 2007, 50, 6700–6705 CrossRef PubMed.
- (a) N. U. Hofsløkken and L. Skattebøl, Acta Chem. Scand., 1999, 53, 258–262 CrossRef; (b) T. V. Hansen and L. Skattebøl, Org. Synth., 2005, 82, 64–68 CrossRef.
- Ø. W. Akselsen and T. V. Hansen, Tetrahedron, 2011, 67, 7738–7742 CrossRef.
- E. J. Solum, A. Vik and T. V. Hansen, Steroids, 2014, 87, 46–53 CrossRef PubMed.
- Ø. W. Akselsen, K. Odlo, J. J. Cheng, G. Maccari, M. Botta and T. V. Hansen, Bioorg. Med. Chem., 2012, 20, 234–242 CrossRef PubMed.
- N. J. Lawrence, A. T. McGown, S. Ducki and J. A. Hadfield, Anti-Cancer Drug Des., 2000, 15, 135–141 Search PubMed.
- R. Abagyan, M. Totrov and D. N. Kuznetsov, J. Comput. Chem., 1994, 15, 488–506 CrossRef.
- R. B. Ravelli, B. Gigant, P. A. Curmi, I. Jourdain, S. Lachkar, A. Sobel and M. Knossow, Nature, 2004, 428, 198–202 CrossRef PubMed.
- Y. Gökmen-Polar, D. Escuin, C. D. Walls, S. E. Soule, Y. Wang, K. L. Sanders, T. M. LaVallee, M. Wang, B. D. Guenther, P. Giannakou and G. W. Sledge Jr, Cancer Res., 2005, 65, 9406–9414 CrossRef PubMed.
- J.-F. Peyrat, J.-D. Brion and M. Alami, Curr. Med. Chem., 2012, 19, 4142–4156 CrossRef PubMed.
- M. P. Leese, H. A. M. Hejaz, M. F. Mahon, S. P. Newman, A. Purohit, M. J. Reed and B. V. L. Potter, J. Med. Chem., 2005, 48, 5243–5256 CrossRef PubMed.
- B. Czako, L. Kurti, A. Mammoto, D. E. Ingber and E. J. Corey, J. Am. Chem. Soc., 2009, 131, 9014–9019 CrossRef PubMed.
- U. Westphal, J. Steroid Biochem., 1983, 19, 1–15 CrossRef CAS PubMed.
- Z. Chen, L. Han, M. Xu, Y. Xu and X. Qian, Curr. Med. Chem., 2013, 20, 1694–1714 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03570h |
| This journal is © The Royal Society of Chemistry 2015 |