
Chemical vapour deposition of Ir-based coatings: chemistry, processes and applications
Abstract
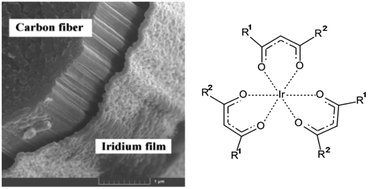
Chemical and materials science aspects of iridium-containing thin film formation by Chemical Vapor Deposition (CVD) methods for modern high-precision technology applications are considered. Chemical approaches to the synthesis of the main precursors used in CVD techniques, thin film growth processes and mechanisms as well as the main structure, composition and properties of iridium-containing thin films are analyzed, and modern thin film application examples are outlined. Numerical characterization of iridium-based thin film growth in 3D objects is presented.
 Please wait while we load your content...
Please wait while we load your content...

