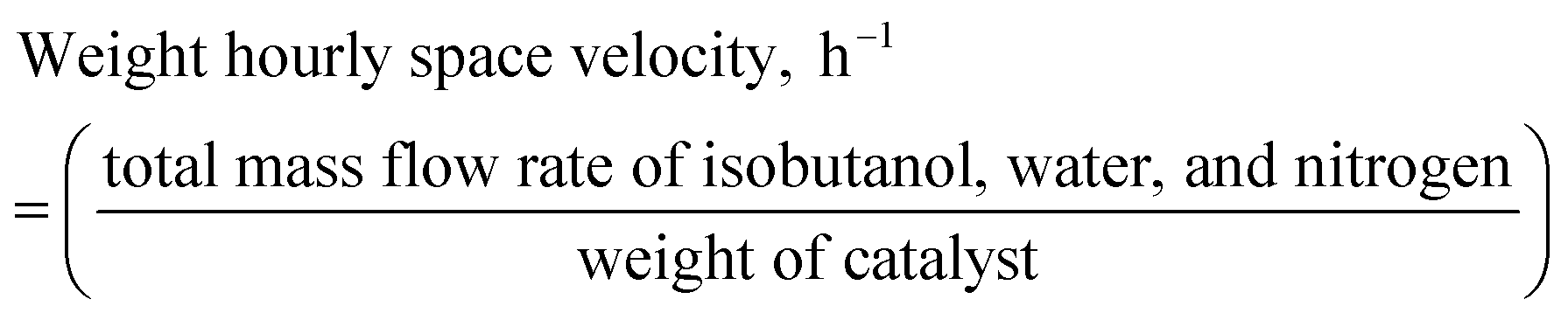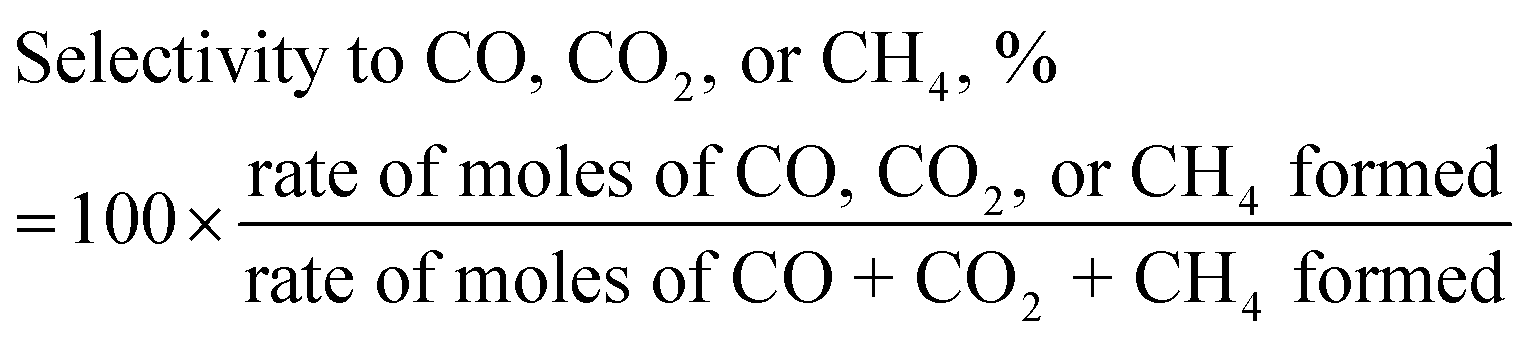DOI:
10.1039/C5RA03558A
(Paper)
RSC Adv., 2015,
5, 52522-52532
Roles of supports (γ-Al2O3, SiO2, ZrO2) and performance of metals (Ni, Co, Mo) in steam reforming of isobutanol†
Received
27th February 2015
, Accepted 8th June 2015
First published on 8th June 2015
Abstract
The production of synthesis gas from bio-isobutanol in an integrated biorefinery is a novel approach for its downstream conversion to hydrocarbon fuels and organic chemicals. The present article provides a systematic examination of the structure–activity correlation of various supported transition metal catalysts, xMS (x mmol metal, M (Ni, Co, and Mo) supported on S (Al, Si, and Zr for γ-Al2O3, SiO2, and ZrO2 respectively)) for steam reforming (SR) of bio-isobutanol. The activity of the catalyst was strongly influenced by metal-support interaction as reflected by metal dispersion, metal crystallite size, and extent of bulk metal/metal oxide. The catalytic activity increased in the order of 4.3NiZr < 4.3NiSi < 4.3NiAl and 4.3MoAl < 4.3CoAl < 4.3NiAl. 7.3CoAl exhibited consistent catalytic activity up to 12 h of time-on-stream. The hydrogen yield was boosted with rise of temperature and steam-to-carbon mole ratio (SCMR) with concurrent drop of selectivity to methane. The selectivity to CO reduced with increasing SCMR and decreasing temperature. Furthermore, spent catalysts were characterized to elucidate the effect of metal and support on the nature of coke formed and chemical transformation of the catalyst during SR.
1 Introduction
The energy and chemical security of the globe is extremely important for sustainability of human civilization. At present our society is heavily dependent on fossil fuels to provide energy, transportation fuels, and organic chemicals. At the moment, more than 80% of energy and greater than 90% of organic chemicals in the world are derived from fossil fuels alone.1 Continuous decline of fossil fuels, escalation of oil prices, and degradation of environmental cleanliness due to large scale usage of fossil fuels forced the exploration of carbon-neutral renewable resources of energy and organic chemicals. Biomass being the origin of fossil fuels has tremendous potential to meet societal needs of both fuels and organic chemicals. At present, ∼10% of total energy or ∼50% of renewable energy comes from biomass alone. Availability of cost-competitive biomass conversion technologies for production of bio-fuels and organic chemicals are thus highly essential to reduce dependence on finite fossil fuels further. Therefore, new manufacturing concepts are continuously emerging to produce an array of bio-fuel and a multitude of bio-products from biomass commonly known as biorefinery.1
The bio-ethanol and biodiesel have been emerged as two promising bio-fuels in the biorefinery with properties suitable for blending with petroleum derived fuels to a limited extent. In recent times, bio-n-butanol has been received renewed attention as bio-fuel due to its superior fuel qualities over bio-ethanol and biodiesel.2–5 These attributes allow direct use of n-butanol in existing internal combustion engine without engine modification. On the other hand, the isobutanol having higher octane number compared to n-butanol and gasoline is considered as a promising bio-fuel of the future.6 Moreover, (n- and iso-) butanol has wide range of market potential as solvent and organic chemicals.7 Once bio-butanol based biorefinery is realized successfully, novel method of production of synthesis gas from butanol must also be established in an integrated biorefinery.
At the moment, almost all organic chemicals manufactured in petrochemical industry are derived from a set of fossil fuels based building blocks. The synthesis gas is one such important petrochemical building block. The synthesis gas provides remarkable opportunities such as production of hydrocarbon fuels and organic chemicals by Fischer–Tropsch synthesis (FTS), raw materials in chemical industries, and source of hydrogen for PEM fuel cell.8–10 The present work was thus commenced on production of synthesis gas by steam reforming (SR) of isobutanol.
Realizing enormous forthcoming potential, several thermodynamic equilibrium analysis were dedicated in the past on SR, dry reforming, partial oxidation, and sorption enhanced SR of butanol to foresee effect of various process parameters on equilibrium product composition and to obtain optimum operating conditions.11–15 However, limited experimental studies are available in open literature on SR of butanol. The SR of n-butanol was studied in presence of co-precipitated Ni/Al2O3 catalyst modified with Cu and Mg and CeO2 and Al2O3 supported nickel catalyst.16–18 SR of ABE mixture (butanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : acetone: ethanol = 6:3: 1) was reported over ZnO, TiO2, and CeO2 supported cobalt catalyst.19 The work was further extended to oxidative steam reforming (OSR) of ABE mixture over ZnO and CeO2–ZrO2 supported cobalt catalyst doped with noble metal (Ru, Rh, Ir, and Pd).20–23 SR of isobutanol was also reported over γ-Al2O3 supported nickel catalyst under wide range of process conditions.11 OSR of isobutanol was investigated over α-Al2O3 supported 1 wt% Rh–1 wt% Ce catalyst in a staged millisecond contact reactor.24 Recently, comparison of experimental OSR results with SR of isobutanol and authentication with equilibrium composition was also reported.25 From the above discussion, it is quite clear that role of support and performance of transition metal for SR of isobutanol was not examined so far. A systematic investigation was therefore undertaken in the present work to provide a comprehensive structure–activity relationship of various inexpensive supported (γ-Al2O3, SiO2, and ZrO2) transition metal (nickel, cobalt, and molybdenum) catalysts for SR of isobutanol. The effect of various process parameters for SR of isobutanol was also investigated over γ-Al2O3 supported cobalt catalysts. The article also provides role of metal and support on nature of coke formation on spent catalyst and chemical transformation of catalyst during SR.
: acetone: ethanol = 6:3: 1) was reported over ZnO, TiO2, and CeO2 supported cobalt catalyst.19 The work was further extended to oxidative steam reforming (OSR) of ABE mixture over ZnO and CeO2–ZrO2 supported cobalt catalyst doped with noble metal (Ru, Rh, Ir, and Pd).20–23 SR of isobutanol was also reported over γ-Al2O3 supported nickel catalyst under wide range of process conditions.11 OSR of isobutanol was investigated over α-Al2O3 supported 1 wt% Rh–1 wt% Ce catalyst in a staged millisecond contact reactor.24 Recently, comparison of experimental OSR results with SR of isobutanol and authentication with equilibrium composition was also reported.25 From the above discussion, it is quite clear that role of support and performance of transition metal for SR of isobutanol was not examined so far. A systematic investigation was therefore undertaken in the present work to provide a comprehensive structure–activity relationship of various inexpensive supported (γ-Al2O3, SiO2, and ZrO2) transition metal (nickel, cobalt, and molybdenum) catalysts for SR of isobutanol. The effect of various process parameters for SR of isobutanol was also investigated over γ-Al2O3 supported cobalt catalysts. The article also provides role of metal and support on nature of coke formation on spent catalyst and chemical transformation of catalyst during SR.
2 Experimental
2.1 Chemicals
Nickel nitrate hexahydrate (purity ≥ 97%) and isobutanol (SG, purity ≥ 99%) were procured from Merck India Ltd., Mumbai. Cobalt nitrate hexahydrate (purity ≥ 98%) and ammonium heptamolybdate tetrahydrate were purchased from Sigma-Aldrich. γ-Al2O3 pellets were procured from Alfa Aesar. SiO2 and ZrO2 pellets were obtained from Saint Gobain NorPro, USA. All chemicals were used without further purification.
2.2 Catalyst preparation
The supported metal catalysts were prepared by multiple incipient wetness impregnation method using respective metal precursor. For each impregnation step, the measured quantity of support (or partially impregnated catalyst) was mixed thoroughly with aqueous metal precursor solution containing requisite amount of metal precursor for about one hour for uniform distribution of metal precursor onto the support. The wet material was then dried in an oven at 373 K for about 16 h followed by calcination at 923 K for about 5 h. These catalysts were hereafter referred as calcined catalyst. After achieving desired metal loading through multiple impregnations, the calcined catalysts were then reduced by hydrogen (20 ml min−1) at 923 K for 3 h. These catalysts were denoted as reduced catalyst throughout the article. The catalyst containing x mmol of metal, M (Ni, Co, or Mo) per gm of support, S (Al, Si, and Zr for γ-Al2O3, SiO2, and ZrO2 respectively) was abbreviated as xMS.
2.3 Catalyst characterization
2.3.1 BET surface area (SA) and pore volume (PV).
SA and PV of the calcined and reduced catalysts together with pure supports were measured using Micromeritics ASAP 2020 physisorption analyzer The samples were first degassed under vacuum (5 × 10−5 mmHg) at 523 K for 2 h. The N2 adsorption and desorption studies were performed at 77 K in the relative pressure (P/P0) range of 0.06 to 0.275. The SA was calculated using multipoint BET equation from adsorption isotherm data. The volume of liquid nitrogen adsorbed at P/P0 = ca. 1.0 was considered as PV.
2.3.2 H2 pulse chemisorption.
H2 pulse chemisorption studies were performed using Micromeritics AutoChem II 2920 chemisorption analyzer to determine metal dispersion (MD) and active metal surface area (SM). The calcined catalyst was first reduced at 923 K using 10 vol% H2–Ar (20 ml min−1) for about 3 h. Sample tube was then purged with argon at a flow rate of 20 ml min−1 for one hour with simultaneous cooling to 323 K to remove traces of hydrogen. Chemisorption studies were then carried out at 323 K by periodical injection of measured volume of H2 pulses until three successive H2 peaks were identical. MD and SM was calculated based on amount of chemisorbed H2 considering surface stoichiometry as H2/M = 0.5.
2.3.3 Powder X-ray diffraction (XRD).
Powder XRD pattern of calcined, reduced, and spent catalysts were acquired in the 2θ range of 10–100° in a Phillips X-pert diffractometer using CuKα radiation (λ = 1.541 Å, 30 kV) with a scanning speed of 0.09° min−1. The metal crystallite size was calculated for different planes by Scherrer's equation using full width half maximum of the XRD peak. The average crystallite size of all the planes was reported in the present article.
2.3.4 Temperature programmed reduction (TPR).
TPR studies of the calcined catalysts were performed in a Micromeritics AutoChem II 2920 chemisorption analyzer equipped with thermal conductivity detector (TCD). Following degassing under the flow of argon (20 ml min−1 at 473 K for about one hour), the sample was cooled down to 323 K. The 10 vol% H2–Ar (10 ml min−1) was then introduced with concurrent rise of temperature from 323 K to 1173 K at a rate of 5 K min−1. The temperature corresponding to maximum hydrogen consumption was considered as maximum reduction temperature (Tmax).
2.3.5 Field emission scanning electron microscopy (FESEM).
FESEM image of the spent catalysts was captured using Zeiss Supra 40 FESEM equipped with energy dispersive X-ray detector.
2.3.6 Fourier transform infrared spectroscopy (FTIR).
FTIR spectra of spent catalysts were recorded using Bruker TENSOR 37 FTIR apparatus equipped with air cooled IR source and low noise DLATGS detector. Spent catalyst was first mixed with KBr and pelletized using hydraulic press. IR spectra were then acquired in transmission mode in the wave number range of 400–4000 cm−1 at ambient temperature with a spectral resolution of 4 cm−1 and 128 number of scan using KBr as background.
2.4 Steam reforming set-up and procedure
SR of isobutanol was carried out under atmospheric pressure in a down-flow fixed-bed reactor. The measured quantity of calcined catalyst pellets (∼3 g) diluted with suitable amount of quartz bead (∼15 g) was loaded into the reactor using quartz wool support. The calcined catalyst was first reduced at 923 K using pure hydrogen with a flow rate of 20 ml min−1 for about 3 h. The reactor was then brought to desired reaction temperature (923 K) under the flow of nitrogen. Controlled volume of isobutanol (0.08 ml min−1) and water (0.25 ml min−1) (isobutanol/water mole ratio of 0.062) were then introduced to a preheater (maintained at 473 K) using two separate HPLC pumps. The vaporized feed from the preheater was then directed to the reactor by controlled flow of nitrogen (53.3 ml min−1). Following liquefaction of condensable fraction in a condenser (265–273 K), the cumulative flow rate of non-condensable product gas was recorded with time-on-stream (TOS) using a wet gas meter. Detailed description of the experimental setup was reported in our earlier publication.11 The gaseous products were quantified by an online gas chromatography (Shimadzu GC 2014) equipped with a TCD using Carbosieve column and argon as carrier gas. The components of the product gas were calibrated using nitrogen as internal standard. The compounds of the liquid samples were identified by a GC attached with a mass spectrometer detector and quantified by an offline GC equipped with flame ionization detector using ZB wax column and helium as carrier gas. The products of liquid sample were also calibrated using HPLC grade pure compounds. Mass balance was checked under steady state for all experiments and error was obtained within ±5%.
3 Results and discussion
3.1 Characterization of the catalysts
3.1.1 Surface area and pore volume.
SA and PV of the catalysts and pure supports are shown in Table 1. The SA and PV of both calcined and reduced catalysts were somewhat lower than respective pure support. The shrinkage of SA and PV might be due to coverage of surface and blockage of pore of support by metal or metal oxide. For γ-Al2O3 supported nickel, cobalt, and molybdenum catalyst with matching mmol of metal loading; the SA and PV increased in the order of 4.3MoAl < 4.3NiAl ∼ 4.3CoAl for both calcined and reduced catalysts. The least SA and PV of 4.3MoAl might be due to higher atomic mass of molybdenum and pore-blockage phenomenon quite often observed in incipient wetness impregnation method. For γ-Al2O3 supported cobalt catalyst; SA and PV reduced continuously with increasing cobalt loading on γ-Al2O3.
Table 1 Physicochemical properties of the catalystsa
| Catalyst |
BET |
Chemisorption |
XRD |
TPR |
| Cal |
Red |
| SA |
PV |
SA |
PV |
MD |
SM |
d
c
|
T
max, K |
|
Cal = calcined; red = reduced; SA = m2 g−1; PV = cm3 g−1; MD = %; SM = m2 g−1 metal; dc = metal crystallite size, nm.
Unreduced forms or oxide forms (MoO3).
|
| γ-Al2O3 |
228 |
0.84 |
— |
— |
— |
— |
— |
— |
| SiO2 |
233 |
0.98 |
— |
— |
— |
— |
— |
— |
| ZrO2 |
38 |
0.2 |
— |
— |
— |
— |
— |
— |
| 4.3NiAl |
178 |
0.6 |
166 |
0.6 |
1.86 |
12.4 |
13.6 |
790, 995 |
| 4.3NiSi |
185 |
0.73 |
143 |
0.7 |
0.29 |
1.90 |
30.9 |
661, 767 |
| 4.3NiZr |
27 |
0.18 |
33 |
0.16 |
0.07 |
0.47 |
26.7 |
661, 681 |
| 4.3MoAl |
119 |
0.49 |
142 |
0.52 |
0.02 |
0.14 |
129.4, 34.9b |
720, 833 |
| 3.0CoAl |
175 |
0.64 |
180 |
0.69 |
0.30 |
2.07 |
15.1 |
775, 874, 942 |
| 4.3CoAl |
166 |
0.58 |
163 |
0.59 |
0.29 |
2.01 |
19.2 |
752, 877, 942 |
| 5.7CoAl |
151 |
0.53 |
149 |
0.53 |
0.18 |
1.22 |
23.4 |
748, 881, 942 |
| 7.3CoAl |
138 |
0.48 |
135 |
0.49 |
0.17 |
1.15 |
60.7 |
745, 885, 942 |
3.1.2 Metal dispersion and metallic surface area.
For γ-Al2O3 supported metal catalysts with equal mmol of metal loading; 4.3NiAl displayed highest MD and SM followed by 4.3CoAl and 4.3MoAl (Table 1). This result clearly demonstrates that nickel has strongest interaction with γ-Al2O3 followed by cobalt and molybdenum. 4.3NiAl exhibited higher MD and SM than 4.3NiSi. The probable role of SA on MD and SM however can be safely nullified as SA of 4.3NiAl and 4.3NiSi are comparable. Therefore, it can be undoubtedly concluded that nickel–γ-Al2O3 interaction is much stronger compared to nickel–SiO2 that leads to higher MD and SM for 4.3NiAl than 4.3NiSi. The 4.3NiZr was however excluded from the comparison of metal-support interaction due to very low SA of ZrO2 as compared to γ-Al2O3 and SiO2. For γ-Al2O3 supported cobalt catalysts; MD and SM decreased steadily with increasing cobalt loading on γ-Al2O3. The decline of MD and SM is due to enrichment of cobalt agglomerates/bulk cobalt with increasing cobalt loading on γ-Al2O3.
3.1.3 Powder XRD.
Powder XRD pattern of calcined catalysts and pure supports with reference to bulk metal oxides are shown in Fig. S1.† Calcined supported nickel oxide catalysts revealed characteristic bulk nickel oxide peaks at 2θ of 37.26° (1 1 1), 43.46° (2 0 0), 62.88° (2 2 0), 75.42° (3 1 1), and 79.5° (2 2 2) [PDF#750197]. The bulk nickel oxide peaks at 2θ of 75.42° and 79.5° were however not observed in calcined 4.3NiAl. Calcined γ-Al2O3 supported cobalt and molybdenum oxide catalyst exhibited representative bulk cobalt oxide (Co3O4) peaks at 2θ of 31.37° (2 2 0), 36.99° (3 1 1), 44.99° (4 0 0), 59.32° (5 1 1), and 65.38° (4 4 0) [PDF#781970] and molybdenum trioxide (MoO3) peaks at 2θ of 20.91° (1 1 1), 23.1° (0 0 2), 23.3° (0 2 0), 23.49° (2 0 0), and 26.3° (2 1 0) [PDF#800347] respectively. From above observations, it may therefore be concluded that calcined catalysts were associated with bulk metal oxide only.
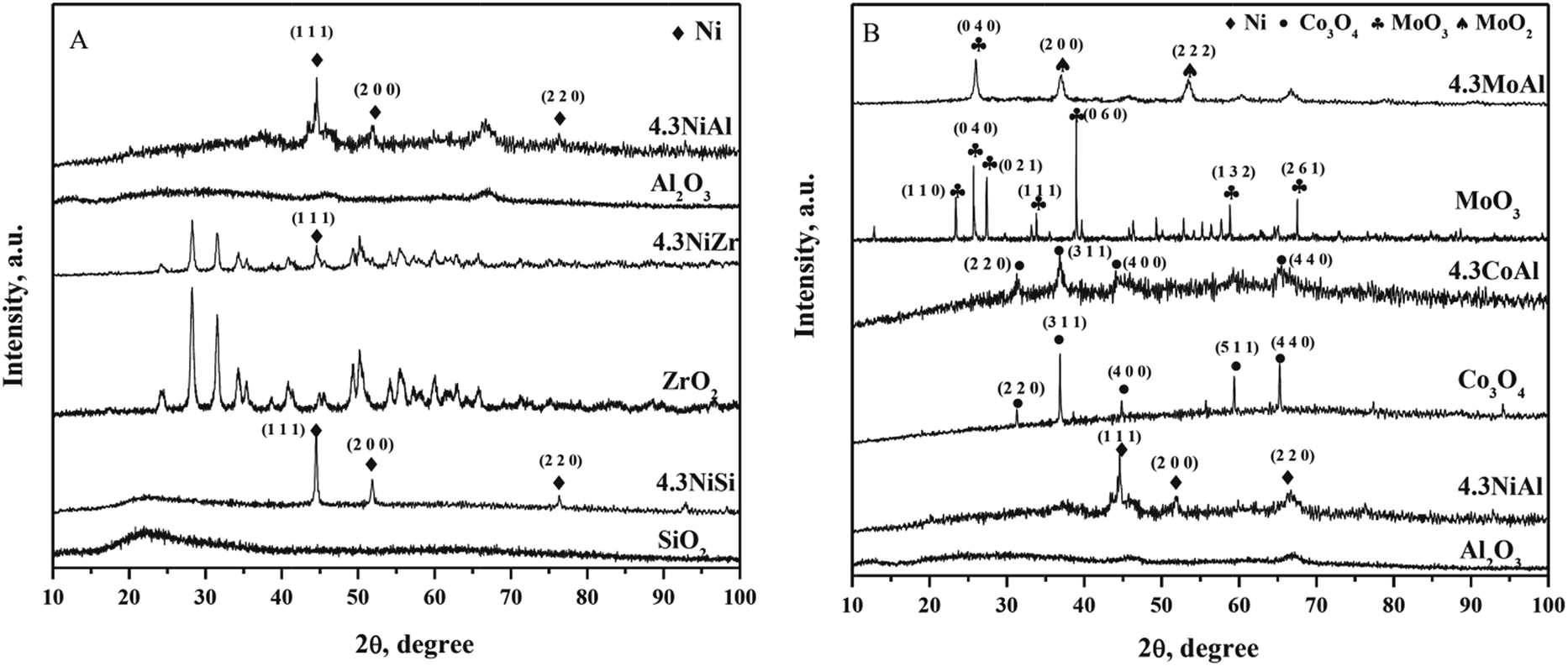
Powder XRD pattern of the reduced catalysts together with pure supports (calcined at 923 K) are shown in Fig. 1. Three characteristic nickel crystallite peaks were identified at 2θ of 44.52° (1 1 1), 51.89° (2 0 0), and 76.44° (2 2 0) for 4.3NiAl, 4.3NiSi, and 4.3NiZr [PDF#701849].26,27 For reduced γ-Al2O3 supported cobalt catalysts; two distinct cobalt crystallite peaks were observed at 2θ of 44.29° (1 1 1) and 75.95° (2 2 0) (PDF#894307). 5.7CoAl and 7.3CoAl however showed an additional Co2AlO4 peak at 2θ of 51.28° (4 0 0). This result clearly suggests that a fraction of cobalt formed solid solution with alumina at elevated cobalt loading on γ-Al2O3. 4.3MoAl however showed peaks corresponding to both molybdenum at 2θ of 40.598° (1 1 0) and 73.74° (2 1 1) (PDF#895156) and molybdenum dioxide (MoO2) at 2θ of 26.19° (1 1 1), 36.89° (2 0 0), and 53.85° (2 2 2) (PDF#761807). The MoO2 peaks were due to incomplete reduction of molybdenum oxides at 923 K. The metal oxide peaks were however not detected in powder XRD pattern of reduced γ-Al2O3, SiO2, and ZrO2 supported nickel and γ-Al2O3 supported cobalt catalysts. This result clearly demonstrates that nickel and cobalt oxide are completely reducible at 923 K.
 |
| | Fig. 1 Powder XRD pattern of reduced catalysts. (A) SiO2, 4.3NiSi, ZrO2, 4.3NiZr, γ-Al2O3, 4.3NiAl, 4.3CoAl, and 4.3MoAl and (B) γ-Al2O3, 3.0CoAl, 4.3CoAl, 5.7CoAl, and 7.3CoAl. | |
Smaller nickel crystallite size for 4.3NiAl compared to 4.3NiSi and 4.3NiZr may be attributed to high SA of γ-Al2O3 and stronger metal-support interaction leading to high MD (Table 1). For reduced γ-Al2O3 supported nickel, cobalt, and molybdenum catalyst; metal crystallite enlarged in the order of Ni < Co < Mo (Table 1). For reduced γ-Al2O3 supported cobalt catalysts; cobalt crystallite enlarged with increasing cobalt loading on γ-Al2O3.
3.1.4 Temperature programmed reduction.
TPR profile of pure supports and calcined catalysts with reference to bulk metal oxides are shown in Fig. 2. γ-Al2O3, SiO2, and ZrO2 showed no reduction peaks thereby confirming that pure supports are fairly stable (thermally) and non-reducible under the range of temperature.28 The calcined 4.3NiAl exhibited two reduction peaks at 790 and 995 K. The lower temperature broad peak represents reduction of bulk nickel oxide having weak interaction with support; whereas higher temperature peak corresponds to reduction of dispersed nickel oxide having strong interaction with γ-Al2O3. The relative peaks area and intensity further suggests that majority of nickel oxide was present in dispersed form in 4.3NiAl.
 |
| | Fig. 2 TPR profile of (A) NiO, SiO2, 4.3NiSi, ZrO2, 4.3NiZr, γ-Al2O3, 4.3NiAl, MoO3, and 4.3MoAl and (B) Co3O4, γ-Al2O3, 3.0CoAl, 4.3CoAl, 5.7CoAl, and 7.3CoAl. | |
On the contrary, calcined γ-Al2O3 supported cobalt and molybdenum oxide catalyst are known to reduce in two separate stages. Three distinct reduction peaks were observed for calcined γ-Al2O3 supported cobalt catalysts at 745–775, 874–885, and 942 K. The first two peaks were associated with reduction of various surface cobalt species; whereas peak at 942 K was due to reduction of CoAlO4 spinel.29 TPR profile of bulk cobalt oxide also showed three different reduction peaks at 642, 679 and 702 K.30 The peak at 679 K was due to reduction of CoO to metallic cobalt; while peaks at 642 and 702 K were due to reduction of Co3O4 to metallic cobalt.31 It was further observed that reduction peaks at 745–775 K and 874–885 K moved progressively to lower and higher temperature respectively with increasing cobalt loading on γ-Al2O3. The shifting of these reduction peaks relative to bulk cobalt oxide peaks may be due to weak interaction of cobalt oxide with γ-Al2O3 and increased extent of different bulk cobalt oxide species with increasing cobalt loading on γ-Al2O3 respectively. Two distinct reduction peaks were observed for calcined 4.3MoAl at 720 and 833 K corresponding to reduction of molybdenum trioxide (MoO3) to MoO2 and MoO2 to molybdenum respectively.
Contrary to calcined 4.3NiAl, bulk and dispersed nickel oxide reduction peaks were observed at lower temperature for calcined 4.3NiSi (661 and 767 K) and 4.3NiZr (661 K and 681 K) (Fig. 2). The relative peaks intensity and area further shows that nickel oxide exists largely in bulk form in calcined 4.3NiZr and 4.3NiSi.31 These results clearly demonstrate that reducibility of supported nickel catalyst depends strongly on nature of support which in turn affects metal-support interaction. From the trend of Tmax (Table 1), it may be further concluded that nickel has strongest interaction with γ-Al2O3 followed by SiO2 and ZrO2.
3.2 Possible steam reforming reactions
SR in general proceeds through large numbers of plausible chemical reactions involving cleavage of carbon–carbon, carbon–oxygen, and carbon–hydrogen bonds leading to formation of wide range of intermediates. For SR of isobutanol, intermediates such as acetaldehyde, propionaldehyde, 2-propenal, butyraldehydes, and 2-butanone together with unreacted (1-, 2-, and iso-) butanol were observed in liquid samples for all experiments (Table 2). Identification and quantification of such intermediates are extremely important to envisage mechanistic pathways for SR of isobutanol. From observed products in liquid samples it may be concluded that isobutanol undergoes isomerization reaction leading to formation of 1- and 2-butanol (Scheme 1). The 1-, 2-, and isobutanol further transformed to butyraldehyde, 2-butanone, and isobutyraldehyde respectively by dehydrogenation reactions. The butanol also undergoes carbon–carbon bond cleavage at different locations leading to formation of various stable intermediates including acetaldehyde, propionaldehyde, and 2-propenal. These intermediates further undergo deep cracking and SR reaction forming H2, CO, CO2, and CH4 as gaseous products (Scheme 2).
Table 2 Carbon balance table for SR of isobutanola
| Catalyst |
Gas products flow rate, mol h−1 |
Liquid products flow rate × 103, mol h−1 |
CBE |
| H2 |
CO |
CH4 |
CO2 |
ACE |
PPD |
PPL |
BUD |
BUN |
BU |
|
ACE = acetaldehyde, PPD = propionaldehyde, PPL = 2-propenal, BUD = (n- and iso-) butyraldehyde, BUN = 2-butanone, BU = 1-, 2-, and isobutanol; CBE = carbon balance error, %.
Isobutanol = 0.052 mol h−1, H2O = 0.83 mol h−1, and N2 = 0.14 mol h−1. Conditions: 923 K, SCMR = 2.2, WHSV = 6 h−1.
Conditions: 7.3CoAl, 923 K, WHSV = 6.5 h−1. Isobutanol flow rate = 0.071, 0.058, 0.052, and 0.039 mol h−1 and H2O flow rate = 0.73, 0.8, 0.9, and 0.86 mol h−1 for SCMR of 1.5, 2, 2.5, and 3.2 respectively, and N2 = 0.14 mol h−1.
Isobutanol = 0.052 mol h−1, H2O = 0.9 mol h−1, and N2 = 0.14 mol h−1. Conditions: 7.3CoAl, SCMR = 2.5, WHSV = 7.02 h−1.
Reproducible results.
|
|
Role of supports
b
|
| 4.3NiAl |
0.474 |
0.064 |
0.017 |
0.122 |
0 |
0.018 |
0.009 |
0.028 |
0.011 |
1.18 |
0.7 |
| 4.3NiSi |
0.427 |
0.063 |
0.014 |
0.105 |
0 |
0.112 |
0.002 |
0 |
0.002 |
1.99 |
7.7 |
| 4.3NiZr |
0.207 |
0.039 |
0.003 |
0.039 |
0 |
1.011 |
0.019 |
0 |
1.174 |
25.04 |
8.4 |
|
|
Performance of metals
b
|
| 4.3NiAl |
0.474 |
0.064 |
0.017 |
0.122 |
0 |
0.018 |
0.009 |
0.028 |
0.011 |
1.18 |
0.74 |
| 4.3CoAl |
0.413 |
0.076 |
0.013 |
0.099 |
0.003 |
0.062 |
0 |
0 |
0.037 |
0.51 |
5.5 |
| 4.3MoA1 |
0.168 |
0.049 |
0.005 |
0.020 |
0.01 |
5.145 |
0.413 |
0 |
0.356 |
24.56 |
7.3 |
|
|
Effect of cobalt loading
b
|
| 3.0CoAl |
0.404 |
0.101 |
0.002 |
0.080 |
0.0285 |
0.497 |
0.08 |
0 |
0.059 |
0.844 |
9.4 |
| 4.3CoAl |
0.413 |
0.076 |
0.013 |
0.099 |
0.003 |
0.062 |
0 |
0 |
0.037 |
0.51 |
5.5 |
| 5.7CoAl |
0.469 |
0.078 |
0.006 |
0.109 |
0.0004 |
0 |
0.01 |
0.0006 |
0.004 |
0.057 |
6.1 |
| 7.3CoAl |
0.463 |
0.073 |
0.010 |
0.115 |
0.001 |
0.093 |
0.01 |
0.093 |
0.005 |
0.051 |
3.5 |
|
|
Effect of steam-to-carbon mole ratio
c
|
| SCMR |
| 1.5 |
0.624 |
0.119 |
0.036 |
0.142 |
0.0008 |
0.0080 |
0.01 |
0.008 |
0.0035 |
0.055 |
3.9 |
| 2 |
0.512 |
0.09 |
0.020 |
0.125 |
0.0003 |
0.0009 |
0 |
0.0009 |
0.0005 |
0.0084 |
1.4 |
| 2.5e |
0.480 |
0.077 |
0.014 |
0.115 |
0 |
0 |
0 |
1.6 × 10−5 |
2.9 × 10−6 |
6.3 × 10−5 |
0.7 |
| 2.5e |
0.464 |
0.073 |
0.011 |
0.115 |
0.001 |
0.0093 |
0.007 |
0.009 |
0.005 |
0.051 |
4.0 |
| 3.2 |
0.420 |
0.047 |
0.004 |
0.101 |
0 |
0 |
0 |
1.0 × 10−5 |
8.3 × 10−6 |
3.2 × 10−5 |
2.2 |
|
|
Effect of temperature
d
|
| 773 K |
0.307 |
0.012 |
0.069 |
0.103 |
0.0008 |
0.0225 |
0 |
0.002 |
0.0016 |
0.93 |
9.7 |
| 823 K |
0.394 |
0.026 |
0.038 |
0.121 |
3.5 × 10−6 |
0 |
0 |
3.3 × 10−5 |
1.5 × 10−6 |
0.827 |
9.5 |
| 873 K |
0.462 |
0.049 |
0.037 |
0.115 |
0.0005 |
0.0001 |
0 |
0 |
0 |
0.003 |
2.8 |
| 923 K |
0.480 |
0.077 |
0.014 |
0.115 |
0 |
0 |
0 |
1.6 × 10−5 |
2.9 × 10−6 |
6.3 × 10−5 |
0.7 |
 |
| | Scheme 1 Plausible chemical reactions during SR of isobutanol. | |
 |
| | Scheme 2 Chemical reactions involved in SR of isobutanol. | |
3.3 Variables for SR of isobutanol
The synthesis gas composition is in general regulated by appropriate adjustment of process variables such as steam-to-carbon mole ratio (SCMR) and weight hourly space velocity (WHSV) as defined below.| |  | (1) |
| |  | (2) |
The stoichiometric SCMR of 7 (eqn (iii) of Scheme 2) was used in the definition of eqn (1). The definition of SCMR signifies extent of excess water supplied as compared to theoretical requirement of one. The catalytic performance was measured in terms of carbon conversion to gaseous products (CCGP), hydrogen yield, and selectivity to CO, CO2, and CH4 as defined below.
| |  | (3) |
| |  | (4) |
| |  | (5) |
As observed from eqn (iii) of Scheme 2, maximum of 12 moles of hydrogen can be produced per mole of isobutanol. In reality, moles of hydrogen produced per mole of isobutanol are far less due to thermodynamic limitation of exothermic water gas shift reaction and incomplete conversion of methane to synthesis gas. Therefore, stoichiometric hydrogen yield of 12 was used in the definition of eqn (4).
The catalytic activity of supported metal catalyst was evaluated for SR of isobutanol under wide range of SCMR (1.5–3.2) and temperature (773–923 K). The molar flow rate of feeds, gaseous, and liquid products were presented in Table 2 for all experiments. The carbon balance error was within ±10% for all experiments. Furthermore, two independent experiments were performed under identical experimental conditions to demonstrate reproducibility of the results for SR of isobutanol (Table 2). The results clearly showed that molar flow rate of products were comparable for both runs thereby demonstrating reproducibility of experimental results.
3.4 Role of supports
For precise comparison of role of supports for SR of isobutanol, three catalysts were prepared with 4.3 mmol of nickel loading on γ-Al2O3, SiO2, and ZrO2. These catalysts were then tested for SR of isobutanol under identical experimental conditions as shown in Fig. 3. As observed from the figure, 4.3NiAl displayed highest catalytic activity with ∼98% CCGP followed by 4.3NiSi with CCGP of ∼88%. The poorer catalytic activity of 4.3NiSi compared to 4.3NiAl might be due to weak metal-support interaction as reflected by its poor MD, bigger nickel crystallite size (Table 1), and larger extent of bulk nickel (Fig. 2). On the other hand, 4.3NiZr demonstrated least catalytic activity with CCGP of only ∼40%. The least catalytic activity of 4.3NiZr might be due to both very low SA (Table 1) and larger extent of bulk nickel (Fig. 2) with poor/or no metal-support interaction. The low MD value of 4.3NiZr also supports above statement (Table 1).
 |
| | Fig. 3 Effect of supports on CCGP, hydrogen yield, and selectivity to CO, CO2, and CH4. All conditions are reported in Table 2. | |
4.3NiAl also showed highest hydrogen yield of ∼76%; while it was ∼68% and ∼33% for 4.3NiSi and 4.3NiZr respectively. The selectivity to methane was however least for 4.3NiZr. The trend of selectivity to CO was totally reverse of trend of CCGP as observed from the figure. This results clearly indicate that water gas shift reaction (eqn (ii) of Scheme 2) become favourable with increasing catalytic activity leading to drop of selectivity to CO and enhancement of CCGP, hydrogen yield, and selectivity to CO2 with increasing catalytic activity.
The unreacted butanols were dominating compound in the liquid sample with insignificant amount of various other intermediates (Table 2). The molar flow rate of butanol and intermediates decreased with increasing CCGP in the order of 4.3NiZr < 4.3NiSi < 4.3NiAl. Since γ-Al2O3 supported nickel catalyst demonstrated superior performance for SR of isobutanol; subsequent studies were performed using γ-Al2O3 as support.
3.5 Performance of nickel, cobalt, and molybdenum
The catalytic performance of γ-Al2O3 supported nickel, cobalt, and molybdenum catalysts with identical mmol of metal loading are shown in Fig. 4. 4.3NiAl showed superior catalytic activity with 98% CCGP followed 4.3CoAl (∼86% CCGP) and 4.3MoAl (∼36% CCGP). The catalytic activity of transition metals for SR of isobutanol was strongly related to their interaction with support. The active metallic site activates oxygenated molecule (e.g. ethanol, isobutanol etc.) and promotes reaction with hydroxyl group generated due to dissociation of water molecule on oxide support. Thus enhancement of catalytic activity was related to enhancement of metal-support interaction (4.3MoAl < 4.3CoAl < 4.3NiAl) that leads to higher MD with smaller metal crystallite size.32 Moreover, presence of a fraction of unreduced molybdenum in 4.3MoAl (Fig. 1) may also be responsible for its inferior catalytic activity.
 |
| | Fig. 4 Effect of metals on CCGP, hydrogen yield, and selectivity to CO, CO2, and CH4. All conditions are reported in Table 2. | |
The hydrogen yield of ∼76% was observed for 4.3NiAl; while it was ∼66% and 27% for 4.3CoAl and 4.3MoAl respectively. The selectivity to CO increased in the order of 4.3NiAl < 4.3CoAl < 4.3MoAl; while trend was opposite for selectivity to CO2. It may be further observed that trend of selectivity to CO was completely reverse of trend of CCGP as observed previously. The similar arguments can also be used to explain the trend of selectivity to CO and CO2. The selectivity to methane was however found to be similar for all the catalysts. The molar flow rate of butanols and intermediates also decreased with increasing CCGP (Table 2). From these results it can be concluded that γ-Al2O3 supported nickel and cobalt catalysts are suitable for SR of isobutanol. The SR of isobutanol over γ-Al2O3 supported nickel catalysts were already reported in our earlier publication.11 The comprehensive study was therefore reported in the present work on SR of isobutanol over γ-Al2O3 supported cobalt catalysts.
3.6 Time-on-stream behavior of 7.3CoAl
The stability of 7.3CoAl was demonstrated for 12 h of TOS as shown in Fig. S2.† The composition of gaseous products reached to steady state within initial ∼100 min of TOS. After 100 min of TOS, variation of composition of gaseous products was insignificant up to 12 h of TOS. Thus γ-Al2O3 supported cobalt catalysts can be considered as fairly stable under the experimental conditions. All reaction data were thus collected after 100 min of TOS.
3.7 Effect of cobalt loading on γ-Al2O3
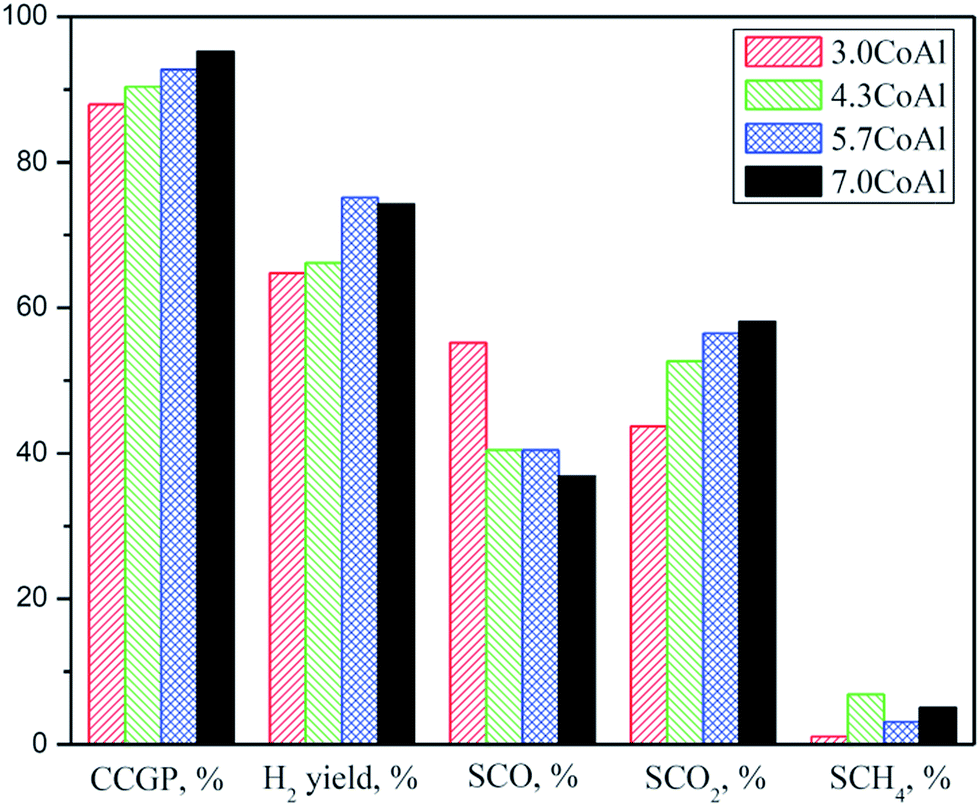
To determine optimum cobalt loading, four different catalysts with 3.0, 4.3, 5.7, and 7.3 mmol of cobalt loading per gm of γ-Al2O3 were tested for SR of isobutanol as shown in Fig. 5. The CCGP enhanced with increasing cobalt loading on γ-Al2O3. About 96% CCGP was observed for 7.3CoAl. For SR of n-butanol over co-precipitated nickel–alumina catalysts, carbon conversion was also reported to increase with increasing nickel loading.18 The hydrogen yield also increased with increasing cobalt loading (up to 5.7 mmol) on γ-Al2O3. The selectivity to CO decreased with increasing cobalt loading on γ-Al2O3; whereas trend was reverse for selectivity to CO2. With increasing cobalt loading on γ-Al2O3, the water gas shift reaction favoured that led to decrease of selectivity to CO and increase of selectivity to CO2 and H2/CO mole ratio. The H2/CO mole ratio in the range of 4.0 to 6.3 was achieved. The effect of cobalt loading on γ-Al2O3 on selectivity to methane was however practically insignificant. Since highest catalytic activity and H2/CO mole ratio was observed for 7.3CoAl, remaining studies were performed over 7.3CoAl.
 |
| | Fig. 5 Effect of cobalt loading on γ-Al2O3 on CCGP, hydrogen yield, and selectivity to CO, CO2, and CH4. All conditions are reported in Table 2. | |
3.8 Effect of steam-to-carbon mole ratio
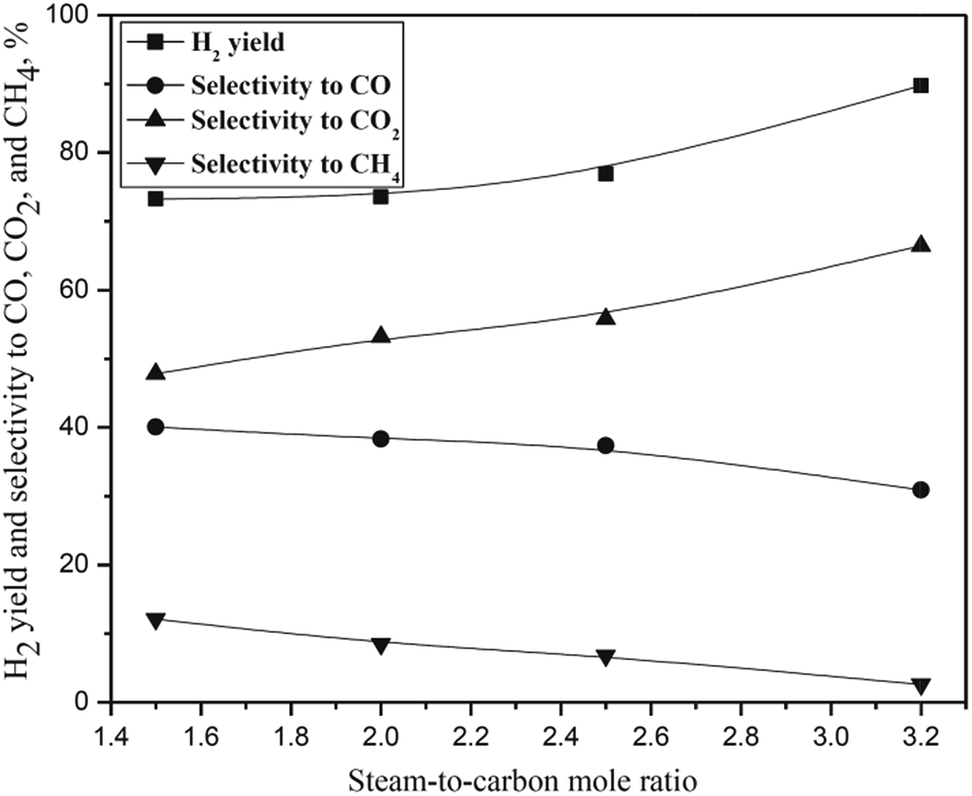
The effect of SCMR was studied in the SCMR range of 1.5–3.2 as shown in Fig. 6. Almost complete CCGP was observed for all runs under the experimental conditions. As observed from the figure, hydrogen yield and selectivity to CO2 increased with increasing SCMR. The hydrogen yield increased from 73% at SCMR of 1.5 to about 90% at SCMR of 3.2. The selectivity to CO and CH4 however decreased with increasing SCMR. Very low selectivity to methane in synthesis gas is highly desirable for its downstream applications for FTS and petroleum or fertilizer industry. As observed from the figure, selectivity to methane decreased from about 12% at SCMR of 1.5 to less than 3% at SCMR of 3.2. The SR of isobutanol (eqn (i) of Scheme 2), intermediate compounds, and methane (reverse of eqn (iv) and (v) of Scheme 2) and water gas shift reaction (eqn (ii) of Scheme 2) are favoured with increasing SCMR that results increasing trend of hydrogen yield and selectivity to CO2 and decreasing trend of selectivity to CO and CH4 with increasing SCMR. It was also observed from Table 2 that mole flow rate of components in liquid sample decreased continually with increasing SCMR.
 |
| | Fig. 6 Effect of steam-to-carbon mole ratio on hydrogen yield and selectivity to CO, CO2, and CH4. All conditions are reported in Table 2. | |
3.9 Effect of temperature
The effect of temperature on SR of isobutanol was studied in the temperature range of 773–923 K as shown in Fig. 7. As observed from the figure, CCGP increased with increasing temperature from merely 25% at 773 K to 100% at 923 K. The endothermic SR reactions (eqn (i) and (iii) of Scheme 2) are favoured at high temperature leading to increasing trend of CCGP with temperature. For SR of n-butanol over co-precipitated nickel–alumina catalysts, carbon conversion was also reported to increase with increasing temperature.18 The hydrogen yield boosted with rise of temperature up to 873 K; beyond which hydrogen yield remained almost unchanged. The selectivity to CO2 and CH4 declined and selectivity to CO increased with increasing temperature. From these results it may be concluded that equilibrium of endothermic SR reactions (isobutanol and methane) (eqn (i), eqn (iii), and reverse of eqn (iv) and eqn (v) of Scheme 2) are favoured at high temperature leading to increase of hydrogen yield and decrease of selectivity to CH4 with increasing temperature. On the other hand, exothermic water gas shift reaction (eqn (ii) of Scheme 2) are favourable at low temperature that results increasing trend of selectivity to CO and decreasing trend of selectivity to CO2 with temperature.
 |
| | Fig. 7 Effect of temperature on CCGP, hydrogen yield, and selectivity to CO, CO2, and CH4. All conditions are reported in Table 2. | |
3.10 Characterization of spent catalysts
The characterization of spent catalyst plays important role to understand nature of catalyst deactivation due to coke formation.33 The possible reactions responsible for coke formation in SR are outlined in Scheme 3. Realizing the significance, several attempts were made in the past to identify nature and amount of coke formed on spent catalyst for SR of various oxygenated compounds.20,22,34,35 In the present work, role of metal and support on nature of coke formed on spent catalysts and chemical transformation of the catalysts during SR of isobutanol were delineated.
 |
| | Scheme 3 Possible coke forming reactions in SR. | |
3.10.1 SEM analysis.
SEM images of spent catalysts are shown in Fig. 8. As observed from the figure, carbon nano-tubes were mainly formed on γ-Al2O3 supported nickel and cobalt catalysts.36 Moreover, carbon nano-tubes grown from the tip of carbon nano-tube containing nickel or nickel carbide particle (white spots in SEM image).37,38 On the other hand, significant amount of carbon spheres, nano-tubes, filaments, and rectangular flakes were observed on spent 4.3MoAl. These observations clearly demonstrated that shape and quantity of carbon formed on spent catalyst is strongly influenced by nature of metal. However, only carbon nano-tubes were observed on spent γ-Al2O3, SiO2, and ZrO2 supported nickel catalysts. The carbon formed a dense nano-tubes network on spent 4.3NiZr; whereas it was dispersed nano-tubes on spent 4.3NiSi. From these results, it can be further concluded that nature of support affects only quantity of coke formed on the spent catalyst without affecting shape of the carbon much.
 |
| | Fig. 8 SEM image of spent (A) 4.3NiAl, (B) 4.3CoAl, (C) 4.3MoAl, (D) 4.3NiSi, and (E) 4.3NiZr. SR conditions: 923 K, SCMR = 2.2, WHSV = 6.62 h−1. | |
3.10.2 Powder XRD.
Powder XRD pattern of selected spent catalysts were acquired without any pretreatments as shown in Fig. 9. The XRD pattern of spent γ-Al2O3, SiO2 and ZrO2 supported nickel catalysts showed features of nickel and respective support only. On the contrary, powder XRD pattern of spent γ-Al2O3 supported cobalt and molybdenum catalyst exclusively exhibited characteristic peaks of cobalt and molybdenum oxide respectively. The Co3O4 peaks were observed at 2θ of 31.37 (2 2 0), 36.99 (3 1 1), 44.99 (4 0 0), 59.32 (5 1 1), and 65.38 (4 4 0) [PDF#781970]. In case of spent 4.3MoAl, both molybdenum trioxide (2θ of 25.88 (0 4 0)) and molybdenum dioxide (2θ of 36.99 (2 0 0) and 53.43 (2 2 2)) peaks were detected [PDF#895108 & PDF#761807]. These results clearly suggested that cobalt and molybdenum was oxidized during SR of isobutanol and vice versa. Therefore, it can be concluded that catalytic activity of cobalt and molybdenum may also depends on rate of oxidation–reduction cycle of metal–metal oxide during SR of isobutanol.
 |
| | Fig. 9 Powder XRD pattern of spent (A) 4.3NiAl, 4.3NiZr, and 4.3NiSi and (B) 4.3NiAl, 4.3CoAl, and 4.3MoAl. SR conditions: 923 K, SCMR = 2.2, and WHSV = 6.62 h−1. | |
3.10.3 FTIR spectroscopy studies.
FTIR spectra of spent catalysts were collected under ambient condition without any further treatment as shown in Fig. S3.† The IR bands appeared at ∼2924 and ∼2850 cm−1 for all spent catalysts were assigned to C–H (νC–H) bond vibration of aliphatic group.39,40 The IR bands observed for spent 4.3NiAl and 4.3NiZr at ∼2960–2970, 2874, 1470–1480, 1410–1420, and 1360–1370 cm−1 (ν(CH), νas(COO−), δ(CH) and νs(COO−)) were due to adsorbed formate species. The IR bands at 1560–1590 cm−1 together with ∼1470–1480 and 1360–1370 cm−1 (νas(COO−), νs(COO−) and δs(CH3)) were due to adsorbed acetate species. A strong IR band observed at ∼1630 cm−1 was assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond vibration (νs(CC)).39,40 The evolution of IR band for CC bond vibration proves presence of unsaturated hydrocarbon precursor on the catalysts which are responsible for formation of carbonaceous deposits.
C bond vibration (νs(CC)).39,40 The evolution of IR band for CC bond vibration proves presence of unsaturated hydrocarbon precursor on the catalysts which are responsible for formation of carbonaceous deposits.
4 Conclusions
The nature of metal and support strongly influenced metal-support interaction which in turn influences catalytic activity for SR of isobutanol significantly. 4.3NiAl (∼98% CCGP) exhibited highest catalytic activity followed by 4.3NiSi (∼88% CCGP), 4.3CoAl (∼86% CCGP), 4.3NiZr (∼39% CCGP), and 4.3MoAl (∼36% CCGP). 7.3CoAl remained fairly stable up to 12 h of TOS. The CCGP enhanced with increasing cobalt loading on γ-Al2O3 with simultaneous reduction of selectivity to CO. With increasing temperature and SCMR, hydrogen yield enhanced with concomitant decrease of selectivity to methane. The selectivity to CO dropped with increasing SCMR and decreasing temperature. The shape and quantity of carbon formed on spent catalyst depends strongly on nature of metal. The XRD pattern of spent catalysts showed that cobalt and molybdenum transformed to oxide during SR of isobutanol.
Nomenclature
| CCGP | carbon conversion to gaseous products, % |
| MD | metal dispersion, % |
| OSR | oxidative steam reforming |
| PV | pore volume, cm3 g−1 |
| SA | BET surface area, m2 g−1 |
| SCMR | steam-to-carbon mole ratio |
| SM | metallic surface area, m2 g−1 metal |
| SR | steam reforming |
|
T
max
| maximum reduction temperature, K |
| TOS | time-on-stream |
| WHSV | weight hourly space velocity, h−1 |
|
xMS | catalyst with x mmol of metal, M (Ni, Co, and Mo) supported on S (γ-Al2O3, SiO2, and ZrO2) |
Acknowledgements
The authors gratefully acknowledge the financial support from Council of Scientific & Industrial Research (CSIR), India (CSIR Ref. no. 22 (673)/14/EMR-II dated 29th December 2014).
Notes and references
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1427–1445 CrossRef CAS PubMed.
- P. Dürre, Curr. Opin. Biotechnol., 2011, 22, 331–336 CrossRef PubMed.
- E. M. Green, Curr. Opin. Biotechnol., 2011, 22, 337–343 CrossRef CAS PubMed.
- B. G. Harvey and H. A. Meylemans, J. Chem. Technol. Biotechnol., 2011, 86, 2–9 CrossRef CAS PubMed.
- M. Kumar and K. Gayen, Appl. Energy, 2011, 88, 1999–2012 CrossRef CAS PubMed.
- C. Cooney, T. Wallner, S. McConnell, J. C. Gillen, C. Abell, S. A. Miers and J. D. Naber, ASME Intern. Combust. Engine Div. Spring Tech. Conf., 2009, 157–165, DOI:10.1115/ICES2009-76155.
- S. K. Maity, Renewable Sustainable Energy Rev., 2015, 43, 1446–1466 CrossRef CAS PubMed.
- P. Biswas and D. Kunzru, Chem. Eng. J., 2008, 136, 41–49 CrossRef CAS PubMed.
- S. R. Yenumala and S. K. Maity, Int. J. Hydrogen Energy, 2011, 36, 11666–11675 CrossRef CAS PubMed.
- S. R. Yenumala and S. K. Maity, J. Renewable Sustainable Energy, 2012, 4, 043120 CrossRef PubMed.
- V. Dhanala, S. K. Maity and D. Shee, RSC Adv., 2013, 3, 24521–24529 RSC.
- G. A. Nahar and S. S. Madhani, Int. J. Hydrogen Energy, 2010, 35, 98–109 CrossRef CAS PubMed.
- A. L. da Silva and I. L. Müller, Int. J. Hydrogen Energy, 2011, 36, 2057–2075 CrossRef PubMed.
- W. Wang and Y. Cao, Int. J. Hydrogen Energy, 2010, 35, 13280–13289 CrossRef CAS PubMed.
- W. Wang and Y. Cao, Int. J. Hydrogen Energy, 2011, 36, 2887–2895 CrossRef CAS PubMed.
- F. Bimbela, D. Chen, J. Ruiz, L. García and J. Arauzo, Appl. Catal., B, 2012, 119–120, 1–12 CrossRef CAS PubMed.
- B. Roy, H. Sullivan and C. A. Leclerc, J. Power Sources, 2014, 267, 280–287 CrossRef CAS PubMed.
- F. Bimbela, M. Oliva, J. Ruiz, L. García and J. Arauzo, J. Anal. Appl. Pyrolysis, 2009, 85, 204–213 CrossRef CAS PubMed.
- W. Cai, P. R. de la Piscina and N. Homs, Bioresour. Technol., 2012, 107, 482–486 CrossRef CAS PubMed.
- W. Cai, N. Homs and P. R. de la Piscina, Green Chem., 2012, 14, 1035–1043 RSC.
- W. Cai, P. R. de la Piscina, K. Gabrowska and N. Homs, Bioresour. Technol., 2013, 128, 467–471 CrossRef CAS PubMed.
- W. Cai, P. R. de la Piscina and N. Homs, Appl. Catal., B, 2014, 145, 56–62 CrossRef CAS PubMed.
- W. Cai, N. Homs and P. R. de la Piscina, Appl. Catal., B, 2014, 150–151, 47–56 CrossRef CAS PubMed.
- R. Chakrabarti, J. S. Kruger, R. J. Hermann and L. D. Schmidt, RSC Adv., 2012, 2, 2527–2533 RSC.
- V. Dhanala, S. K. Maity and D. Shee, J. Ind. Eng. Chem., 2015, 27, 153–163 CrossRef CAS PubMed.
- M. Benito, R. Padilla, L. Rodríguez, J. L. Sanz and L. Daza, J. Power Sources, 2007, 169, 167–176 CrossRef CAS PubMed.
- L. Wang, D. Li, M. Koike, H. Watanabe, Y. Xu, Y. Nakagawa and K. Tomishige, Fuel, 2013, 112, 654–661 CrossRef CAS PubMed.
- J. D. A. Bellido and E. M. Assaf, J. Power Sources, 2008, 177, 24–32 CrossRef CAS PubMed.
- H. F. J. van 't Blik and R. Prins, J. Catal., 1986, 97, 188–199 CrossRef.
- V. A. de la Peña O'shea, R. Nafria, P. R. de la Piscina and N. Homs, Int. J. Hydrogen Energy, 2008, 33, 3601–3606 CrossRef PubMed.
- J. Zhu, X. Peng, L. Yao, J. Shen, D. Tong and C. Hu, Int. J. Hydrogen Energy, 2011, 36, 7094–7104 CrossRef CAS PubMed.
- S. Li, C. Zhang, Z. Huang, G. Wu and J. Gong, Chem. Commun., 2013, 49, 4226–4228 RSC.
- B. M. Güell, I. Babich, K. P. Nichols, J. G. E. Gardeniers, L. Lefferts and K. Seshan, Appl. Catal., B, 2009, 90, 38–44 CrossRef PubMed.
- A. L. Alberton, M. M. V. M. Souza and M. Schmal, Catal. Today, 2007, 123, 257–264 CrossRef CAS PubMed.
- X. Hu and G. Lu, Appl. Catal., B, 2010, 99, 289–297 CrossRef CAS PubMed.
- J. Vicente, C. Montero, J. Ereña, M. J. Azkoiti, J. Bilbao and A. G. Gayubo, Int. J. Hydrogen Energy, 2014, 39, 12586–12596 CrossRef CAS PubMed.
- I. E. Achouri, N. Abatzoglou, C. Fauteux-Lefebvre and N. Braidy, Catal. Today, 2013, 207, 13–20 CrossRef CAS PubMed.
- M. N. Barroso, A. E. Galetti and M. C. Abello, Appl. Catal., A, 2011, 394, 124–131 CrossRef CAS PubMed.
- D. Shee and G. Deo, J. Mol. Catal. A: Chem., 2009, 308, 46–55 CrossRef CAS PubMed.
- D. Shee and A. Sayari, Appl. Catal., A, 2010, 389, 155–164 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Powder XRD pattern of calcined catalysts; time-on-stream behavior of 7.3CoAl catalyst; FTIR profile of spent catalysts. See DOI: 10.1039/c5ra03558a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence