
Optimized syringe-assisted dispersive micro solid phase extraction coupled with microsampling flame atomic absorption spectrometry for the simple and fast determination of potentially toxic metals in fruit juice and bio-fluid samples†
Behruz Barfi,
Alireza Asghari*,
Maryam Rajabi,
Sedigheh Sabzalian,
Forough Khanalipoor and
Mahdi Behzad
Department of Chemistry, Semnan University, Semnan 35195-363, Iran. E-mail: aasghari@semnan.ac.ir; Fax: +98-231-3354110
First published on 27th March 2015
Abstract
In this work, a novel method called Syringe-assisted dispersive micro solid phase extraction (SA-DM-SPE) was developed based on repeatedly withdrawing and pushing out a mixture of an aqueous sample including some chelated potentially toxic metal ions with bis-(acetylacetone) ethylenediimine and a low level of a suitable adsorbent (1.6 mg of multi-walled carbon nanotubes) in a test tube using a syringe. Since maximum contact surface areas were simply provided between the chelated ions and adsorbent with no need to essentially off-line the accelerating mass transfer (including sonication and vortex) and centrifugation steps, maximum efficiency was achieved within a short period of time. The optimized conditions for the extraction of Pb2+, Cd2+, Co2+, Ni2+, and Cr3+, as target ions, were investigated by the experimental design strategy. Under the optimum conditions, limits of detection, linear dynamic ranges, consumptive indices, and repeatabilities (in terms of intra-day precisions) ranged from 0.3 to 2.0 μg L−1, 0.9 to 980 μg L−1, ∼0.33, and 3.4 to 4.2, respectively. The method was successfully applied to the determination of target ions in different water (tap and wastewater), fruit juice (apple, pear, grape, and grapefruit), and biological fluid (saliva and urine) samples using a microsampling flame atomic absorption spectrometry (MS-FAAS) technique.
1. Introduction
Among the environmental pollutants, potentially toxic metal ions generate the greatest concern to general public health, and therefore, are very important to the environmental agencies in most countries. The main sources of continuous release of these metals are industrial and agricultural activities, combustion of fossil fuels, and atmospheric emissions.1,2 Food and water are the two main sources that can transfer the potentially toxic metal ions to the human body. Consumption of food and water with high concentrations of these metals can produce a variety of problems for the human health such as depletion of immunological defenses, intrauterine growth retardation, disabilities associated with malnutrition, and a high prevalence of upper gastrointestinal cancer. In this way, it is of great importance to develop simple and efficient methods for the determination of trace potentially toxic metals in biological, nutritional, and environmental samples.3–5Several different techniques such as flame atomic absorption spectrometry (FAAS), electro-thermal atomic absorption spectrometry (ETAAS), inductively coupled plasma-optical emission spectrometry (ICP-OES), inductively coupled plasma-mass spectrometry (ICP-MS), and electrochemical-based methods have been frequently used for the determination of potentially toxic metals in various real samples.6–10 Among them, FAAS has been frequently applied for metal ion monitoring in different real samples due to its low cost, operational facility, and high sample throughput. Despite these advantages as well as the matrix complexity of real samples, some metals have low concentrations near or below the detection limit of this technique. Under these circumstances, a separation and enrichment step can be beneficial prior to their trace determination. However, in comparison with ETAAS and ICP-OES, a relatively large volume of the eluent is needed for multi-element analysis by FAAS, which leads to decrease in the enrichment factor and sensitivity of the technique. To overcome this drawback, microsampling with the aid of home-made devices can be a good solution. In the microsampling-FAAS, a small volume of the eluent is pipetted into a Teflon funnel, and directly nebulized by a conventional capillary pneumatic nebulizer in a premixed flame.11 The responses are recorded in terms of the peak areas and depicted precision and sensitivity, similar to those obtained with a normal larger (1–5 mL) eluent by FAAS.12 This approach was applied in the present work, and 300 μL of the eluent proved to be sufficient for the determination of five potentially toxic metals in different real samples.
Modern trends in analytical chemistry are towards the miniaturization and simplification of sample preparation (especially for extraction methods) as well as minimizing the extractant phase along with a high enrichment and clean-up. In order to achieve these purposes, various extraction and microextraction methods such as solid phase extraction (SPE),5,13 dispersive-solid phase extraction (D-SPE),14–16 matrix solid phase dispersion (MSPD) extraction,17,18 membrane extraction (ME),19 stir-bar sorptive extraction (SBSE),20 solid phase microextraction (SPME),21 and liquid phase microextraction (LPME)6,22–25 have been developed.
D-SPE is a modified version of SPE that considerably reduces the time consumed, and simplifies the extraction process. In this method, extraction is not carried out in a cartridge, column or disk but in the bulk solution, which leads to more rapidity and ease of operation compared with the conventional SPE. The method consists of two critical steps: (i) dispersion, and (ii) phase separation. The first step is usually assisted by an external energy source, and therefore, special apparatus such as ultrasonic and vortex are required. Although the use of organic solvents has also been proposed for dispersion, these substances may enhance the solubility of target analytes in the sample, and thus reduce the extraction efficiency.26 The second step is usually performed by centrifugation, which is very effective. However, it makes the overall procedure time-consuming. In this sense, development of a D-SPE method which could avoid the use of external apparatus and even organic solvents, without centrifugation, is of great importance (especially for the on-site extraction in environmental analysis).27,28 When few amounts of the adsorbent (at very low mg ranges) are used, the method is called dispersive micro solid phase extraction (DM-SPE).
So far, various adsorbents have been utilized to trap or adsorb the target analytes in different real samples.29–31 The nature and properties of the adsorbent are of prime importance in DM-SPE. In practice, the main requirements for an adsorbent are: (i) fast adsorption, (ii) quantitative recovery, and (iii) high surface area, capacity, and dispersibility in liquid samples. In this context, magnetic and carbonaceous nanomaterials seem to be perfect for use in this method. Carbon nanotubes (CNTs) are novel and interesting carbonaceous materials, which are classified as single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) on the principle of presence of carbon atom layers in the walls of nanotubes.32 Due to their remarkable physical and chemical properties, MWCNTs have attracted increasing interest as sorbents for the SPE methods. However, to the best of our knowledge, there are a few reports on the application of MWCNTs (with or without modifications) as adsorbents for DM-SPE of potentially toxic metals in real matrices.33
In the present study, the simple, fast, efficient, and optimized syringe-assisted DM-SPE (SA-DM-SPE) method was developed to determine the Pb2+, Cd2+, Co2+, Ni2+, and Cr3+ ions, as model analytes, in different biological fluid (saliva and urine), fruit juice (apple, orange, pear, grape and grapefruit), and water (tap and wastewater) samples using a microsampling flame atomic absorption spectrometry (MS-FAAS) technique. To achieve the best extraction efficiency, the effective parameters were investigated and optimized by the central composite design.
2. Experimental
2.1. Instrumentation
All the measurements were performed with an Agilent 200 Series AA (model 240 AA) flame atomic absorption spectrometer (USA) including air–acetylene flame and simultaneous four hollow cathode lamps. The instrumental parameters were adjusted as follow: wavelength Pb 217.0 nm (slit width: 1.0 nm), Cd 228.8 nm (slit width: 0.5 nm), Co 240.7 nm (slit width: 0.2 nm), Cr 357.9 nm (slit width: 0.2 nm), Ni 232.0 nm (slit width: 0.2 nm), and lamp current: 10.0 mA. The eluent phase (300 μL), 60 μL for each ion, was taken and injected into the FAAS nebulizer using a home-made microsample introduction system consisting of a Teflon funnel and an Eppendorf pipette, and the peak areas were measured. The pH values for the solutions were measured using a PHS-3BW model pH-meter (Bell, Italy).2.2. Reagents and solutions
The acids, bases, and other solvents used were of the highest purity, available from Merck (Darmstadt, Germany, http://www.merck.de). Nitrate salts of all the metal ions including analytes and interferences, purchased from Merck, were of the highest purity. Stock solutions (1000 mg L−1) of all the ions under study were prepared by dissolving appropriate amounts of their salts in nitric acid (2 mol L−1). The working standard solutions used for calibration were prepared by appropriate dilutions of the stock standard solutions with doubly distilled water. The calibration standards were subjected to the microextraction method. The chelating agent bis-(acetylacetone)ethylenediimine (BAAED) was synthesized in the laboratory.34 A solution of BAAED (0.10 mol L−1) was prepared by dissolving an appropriate amount of this chelating agent in ethanol. The adsorbent (MWCNTs; purity > 95%) with diameters of 6–9 nm and lengths of ca. 5 μm were purchased from Sigma-Aldrich (St. Louis, MO, USA, http://www.sigmaaldrich.com). CRM-TMDW-100 (drinking water standard) (High-Purity Standards, http://www.highpuritystandards.com), and NIST SRM 1640a (natural water standard) (National Institute of Standards and Technology, http://www.nist.gov) were used to check the accuracy of the proposed method. Diluted nitric acid and sodium hydroxide solutions were used for the adjustment of the pH value to the desired one. The vessels used for trace analysis were kept in 10.0% nitric acid for at least 24 h, and subsequently washed with distilled water.2.3. Sample preparation
(i) Do not take vitamins or aggregated minerals 36 h before the saliva or urine collection.
(ii) Exclude brushing teeth before the saliva collection.
(iii) Avoid chewing gum for at least 12 h before the collection.
(iv) Remit the collected samples directly to the laboratory for analysis.
2.3.1.1. Human saliva. The saliva samples were taken in the morning before breakfast. The volunteers were asked to rinse their mouth for 1 min using 10 mL of doubly distilled water. Immediately after the rinsing, about 12 mL of unstimulated saliva were collected for 10 min with the mouth closed, and introduced into a number of polyprophylene tubes. The saliva samples were immediately centrifuged at 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 min in order to sediment cellular debris. The patients with orthodontic appliances or samples with visible blood contamination were discarded.35 10 mL of the collected samples were stored at −4 °C before they were subjected to SA-DM-SPE.
000 rpm for 5 min in order to sediment cellular debris. The patients with orthodontic appliances or samples with visible blood contamination were discarded.35 10 mL of the collected samples were stored at −4 °C before they were subjected to SA-DM-SPE.
2.3.1.2. Human urine. Morning urine samples were collected in plastic bottles, and stored at −20 °C till analysis. Before use, the samples were thawed, and the working solutions were prepared into 10 mL volumetric flasks. The urine samples were filtered and subjected to SA-DM-SPE.
2.4. SA-DM-SPE method
1.7 mg of the MWCNTs was added to a 10 mL glass tube, and 10.0 mL of each spiked sample solution (containing 100.0 μg L−1 of each metal ion and 0.07 mol L−1 of ligand) was pipetted into the tube. Using a gas-tight syringe, the mixture was rapidly withdrawn and pushed out into the tube for 10 times within a time duration of 30 s. After extraction, the whole volume of the sample solution and sorbent was aspirated in the syringe, and then filtered through a syringe filter. The filtrate was retracted, and the adsorbent was eluted out with nitric acid solution (3.5 mol L−1) in a time duration of 30 s. The eluent was collected (300 μL), and analyzed using MS-FAAS to determine the metal concentrations (Fig. 1). The absorbance signals were measured as peak areas with a 3 s integration time. | ||
| Fig. 1 Schematic set-up of syringe-assisted dispersive micro solid phase extraction coupled with microsampling flame atomic absorption spectrometry. | ||
2.5. Calculations of enrichment factor, absolute and relative recoveries
The enrichment factor (EF), absolute recovery (extraction recovery, ER), and relative recovery (RR) for the analytes were used as the parameters to evaluate the method. EF was calculated by eqn (1).
 | (1) |
ER was calculated by eqn (2).
 | (2) |
RR was calculated by eqn (3).
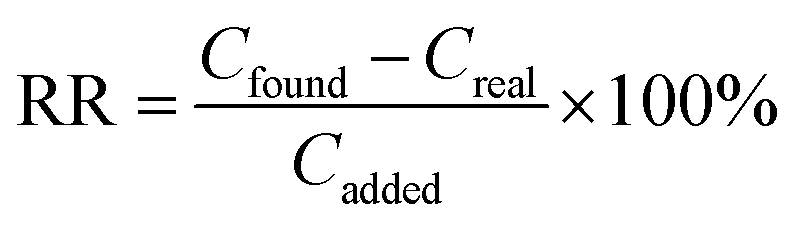 | (3) |
2.6. Central composite design
Central composite design (CCD) is an effective design that is used for sequential experimentation, and provides a reasonable amount of information for testing the goodness of fit. It does not require an unusually large number of design points, and thereby, reduces the overall cost associated with the experiment. By using CCD, the experimenter can start with a model of low order, possibly even a linear model, which is the lowest possible order. If the resulting model does not appear to be adequate, it is possible to simply add new observations to the existing ones and fit a higher-order model, giving new regression coefficients. After concluding that a linear model is inadequate, one can continue the same investigation by adding additional measurements at the star points and in the center. This design was used to optimize the simultaneous effects of parameters including the main, interaction, and quadratic effects on the analyte extraction efficiency. In order to evaluate these effects, thirty-one experiments including sixteen axial points, eight star points, and seven center points were performed in this design. The Design Expert (DE) software (version 7.0.0) was used for the analysis of the experimental design data and calculating the predicted responses.3. Results and discussion
In order to reach a high extraction efficiency (in terms of recovery, R), the influence of different parameters affecting the adsorption step (including type of adsorbent (TA), amount of adsorbent (AA), concentration of ligand (CL), pH of sample, and number of extraction cycles (NEC)) as well as factors affecting the desorption step (including type of eluent (TE), volume of eluent (VE), and concentration of eluent (CE)) were investigated.Before application of CCD, preliminary experiments were undertaken to select the best type of adsorbent and desorption conditions using the one-variable-at-a-time (OVAT) design. To this end, the SA-DM-SPE method was applied for extraction of 100 μg L−1 of the spiked ions from the sample solutions.
3.1. Type of adsorbent
Careful attention should be paid in the selection of the adsorbent. The extraction process usually involves adsorption of the metal ions at the surface of the adsorbent via the interactions with various functional groups, chelation, and ion-pair formation processes. The mechanism of analyte adsorption on a solid phase depends upon the nature of the adsorbent and its interaction with the chelated ions. Compared with the ordinary adsorbents, nano-sized sorbents demonstrate higher surface areas. Therefore, satisfactory results can be achieved by lower amounts of these adsorbents. In this way, the extraction efficiencies of 1.5 mg of different adsorbents such as zinc oxide (ZnO), activated carbon (AC–) modified with tin sulfide (SnS), ruthenium (Ru), and gold (Au) nanoparticles, all synthesized in our laboratory, were compared with the MWCNTs. The results obtained show that MWCNTs provide a better adsorption efficiency for the analytes (in terms of the extraction recovery) compared with the other adsorbents (Fig. 1S) (ESI†). Hence, further experiments were followed with this adsorbent.3.2. Type, concentration, and volume of eluent
For desorption of the metal–chelate complexes from MWCNTs, a series of selected eluent solutions such as HNO3, HCl, CH3COOH, and H2SO4 were used at the pre-determined adsorption conditions including TA: MWCNTs, AA: 2 mg, CL: 0.1 mol L−1, pH: 6.5, NEC: 15, and equal eluent concentrations (2.0 mol L−1). The results obtained showed that HNO3 provided more effective elution of the target ions from the adsorbent.The eluent concentration was studied in the range of 1.0 to 5.0 mol L−1. The best results were achieved when 3.5 mol L−1 of HNO3 was used as the eluent. Therefore, this concentration was used to achieve the best recoveries.
Selection of the elution conditions was continued in order to obtain the maximum recovery with a minimum volume of the eluent. Although at the eluent volumes lower than 300 μL (at 250 μL) the recovery of the ions was quantitative, satisfactory results were not obtained due to insufficient repeatabilities. The results obtained revealed that 300 μL of HNO3 solution (3.5 mol L−1) was the best elution condition for the subsequent experiments.
3.3. Central composite design
The central composite design was applied for examination of the interactions between the variables involved in the adsorption step. Random experiments were conducted to minimize the effects of uncontrolled variables and conditions, and the results obtained were tabulated in Table 1.| Factors | Levels | Starpoint α = 1.682 | |||
|---|---|---|---|---|---|
| Low | Central | High | −α | +α | |
| Amount of adsorbent (AA) (mg) | 0.75 | 1.38 | 2.00 | 1.06 | 1.69 |
| Concentration of ligand (CL) (mol L−1) | 0.00 | 0.05 | 0.10 | 0.03 | 0.08 |
| pH of sample (pH) | 2.00 | 5.5 | 9.00 | 3.75 | 7.25 |
| Number of extraction cycles (NEC) | 5.00 | 9.00 | 13.00 | 7.00 | 11.0 |
In order to find the most important effects and interactions, analysis of variance (ANOVA) was performed using the DE software (Table 2). The statistical significance of all the terms in the model was tested by the F-value and P-value. The corresponding variables would be more significant if the P-value of lack of fits (LOF) became greater than 0.05, and the P-value of regressions became smaller than 0.5. An F-value greater than 35.66 implies that the model is statistically significant, and there is only a 0.01% chance that the “F-value model” is due to noise.
| Analytes | Lack of fit | Regression coefficients | |||
|---|---|---|---|---|---|
| P-value regression | P-value lack of fit | F-valuea | R2 | Radj2 | |
| a Model F-value. | |||||
| Pb2+ | <0.001 | 0.2413 | 43.17 | 0.9401 | 0.9183 |
| Cr3+ | <0.001 | 0.1639 | 35.66 | 0.9469 | 0.9203 |
| Ni2+ | <0.001 | 0.2851 | 46.96 | 0.9527 | 0.9324 |
| Cd2+ | <0.001 | 0.1516 | 40.25 | 0.9360 | 0.9128 |
| Co2+ | <0.001 | 0.0914 | 36.18 | 0.9476 | 0.9214 |
The regression coefficients including the determination coefficients (R2) and adjusted determination coefficients (Radj2) were used to estimate the goodness of the fit of the model; they are listed in Table 2. The R2 values were greater than 0.9360, which indicated that 6.4% of the variations could be explained by the predicted model. The Radj2 values greater than 0.9128 indicated good degrees of correlation between the observed and predicted values. Both values ensured a satisfactory adjustment of the polynomial model to the experimental data.
Data analysis gave the semi-empirical expressions of the extraction recovery for the chelated ions, as follow:
| R(Pb2+) = −163.15 + 128.49*AA + 606.83*CL + 16.11*pH + 17.59*NEC − 38.80*(AA)2 − 4264.25*(CL)2 − 1.39*(pH)2 − 0.85*(NEC)2 | (4) |
| R(Cr3+) = −147.88 + 151.86*AA + 285.54*CL + 13.25*pH + 11.57*NEC − 256.90*AA*CL + 80.13*CL*pH − 44.21*(AA)2 − 2555.34*(CL)2 − 1.34*(pH)2 − 0.52*(NEC)2 | (5) |
| R(Ni2+) = −146.71 + 126.57*AA + 173.68*CL + 22.47*pH + 9.56*NEC + 35.70*CL*NEC − 39.52*(AA)2 − 3622.29*(CL)2 − 1.69*(pH)2 − 0.55*(NEC)2 | (6) |
| R(Cd2+) = −128.73 + 87.64*AA + 606.81*CL + 20.48*pH + 14.72*NEC − 27.19*(AA)2 − 5008.40*(CL)2 − 1.69*(pH)2 − 0.71*(NEC)2 | (7) |
| R(Co2+) = −99.87 + 17.72*AA + 987.04*CL + 19.92*pH + 15.10*NEC − 319.23*AA*CL + 6.73*AA*pH − 11.73*(AA)2 − 4215.79*(CL)2 − 2.29*(pH)2 − 0.75*(NEC)2 | (8) |
The models are applicable for prediction of the recovery of the analytes with a minimum number of experiments. Typical plots of the predicted vs. the observed response, and the residuals vs. the predicted response are shown in Fig. 2a and b. A close inspection of Fig. 2a reveals that the residuals are generally close to a straight line, which indicates the normal distribution of the error, and supports the fact that the model adequately fits the data. These plots are very important, and it is required to check the normality assumption in the fitted model. This ensures that the model provides an adequate approximation to the optimization process. It is clear that no obvious pattern is followed in the residual vs. the predicted response (Fig. 2b).
 | ||
| Fig. 2 (a) Plot of predicted values vs. observed values for the recovery (%) of Ni2+ ions (b) plot of residuals vs. predicted response for the recovery (%) of Ni2+ ions. | ||
In order to represent the effects of important interactions on the results, the response surface plots including the 3-D and contour plots of the model were prepared using the DE software. These plots also demonstrated the quality of the relation between the recoveries and experimental levels of significant factors. In these plots, the recovery is mapped against two experimental factors, and the remaining factors are usually held constant at their center points. Fig. 3 represents typical 3-D and contour plots of the effects of significant parameters on the Ni2+ recovery.
 | ||
| Fig. 3 Response surfaces for Ni2+ as a representative analyte: (a) concentration of ligand (CL) vs. amount of adsorbent (AA) (pH of sample, and number of extraction cycles (NEC), fixed at 5.5 and 9, respectively); (b) pH vs. AA (CL and NEC, fixed at 0.05 mol L−1 and 9, respectively); (c) NEC vs. AA (pH and CL, fixed at 5.5 and 0.05 mol L−1, respectively); (d) pH vs. CL (AA and NEC, fixed at 1.4 mg and 9, respectively); (e) pH vs. NEC (AA and CL, fixed at 1.4 mg and 0.05 mol L−1, respectively). | ||
The effect of the amount of adsorbent was also studied so as to determine the lowest amount of the adsorbent required to obtain the highest extraction efficiency for the chelated ions. As expected, as the amount of the adsorbent increased, higher recoveries were obtained, and then they remained almost constant with a further increase in the amount (when a constant volume of the sample was used). Evidently, at lower amounts of MWCNTs, the available surface areas were inadequate to afford the quantitative recovery of the target ions (Fig. 3a–c).
The metal–chelate stability constants and their chemical stability significantly influence the analyte recovery. The pH value for the sample has a unique role in this stability and the subsequent extraction efficiencies because it not only affects the formation of metal–chelate complexes but also allows the formation of hydrophobic complexes that can be adsorbed on the MWCNT surfaces through van der Waals forces and hydrophobic interactions (Fig. 3b, d, and e). At a lower pH value (less than 6), the hydroxyl group and nitrogen atom in BAAED are protonated, and thus the extraction efficiency decreases. On the other hand, at pH > 7.1, the recoveries also decrease, and this may be due to the precipitation of some ions in the form of hydroxides.
Concentration of the ligand has a direct effect on the formation of the metal–chelate complexes and their subsequent adsorption on MWCNTs. As it can be seen, with an increase in the amount of ligand, an increase in the recovery can be achieved, and a further increase does not enhance the efficiency (Fig. 3a and d).
The extraction efficiency of Dμ-SPE depends upon the mass transfer velocity of the target analytes from the sample solution to the adsorbent. Due to the high surface area to volume ratios in MWCNTs and their short diffusion routes, which lead to a rapid adsorption process, the equilibrium between the chelated ions in the sample solution and the adsorbent surface can be reached in a short contact time. The dispersion phenomenon could accelerate the possible contact between the adsorbent and the sample solution, and accessible surface areas of the adsorbent are achieved in a shorter period of time. In this way, it is predictable that, by increasing NEC, the recovery should also increase. However, when constant amounts of the adsorbent and sample are used, the recoveries remain constant, after reaching the equilibrium status (Fig. 3c and e).
The desirability function (DF) is a common and established technique to discover the global optimal conditions based on the Derringer's desirability function. DF distinguishes and creates a function for each individual response. Finally, it determines a global function that should be maximum following selection of optimum values of the effective variables, considering their interactions. Fig. 2S† shows the desirability versus the response surfaces of target ions. The scale in the range of 0.0 (undesirable) to 1.0 (very desirable) is used to obtain a global function according to an efficient selection and optimization of the designed variables. On the basis of the evaluations and desirability score (closeness to 1.0), maximum responses were obtained at the optimum conditions including TA: MWCNTs, AA: 1.6 mg, CL: 0.07 mol L−1, pH: 6.4, NEC: 10, TE: HNO3, VE: 300 μL, and CE: 3.5 mol L−1.
3.4. Potentially interfering ions
The competitive or synergistic effect of other cations and anions on the method performance was examined individually. The interference was considered to occur when the measured recoveries varied more than ±5% relative to those for the target ions. In this effort, some model solutions containing 50.0 μg L−1 of the standard mixtures were fortified with increase in the amount of potentially interfering ions, selected on the basis of their common occurrence in real samples. The results indicated that the method could be applied to the real samples containing the target ions since it is not affected by high concentrations of the alkali and alkaline earth ions (up to and other transition metal ions (Table 3). However, some trace coexisting metal ions that effectively compete for complexation with BAAED can interfere and reduce the extraction efficiency. The lowest recoveries were found in the presence of Cu2+ and Zn2+ ions that interfere, at the concentrations 45 times more than those for the ions under study.| Ion | Concentration (mg L−1) | Added as | Mass ratioa | Recovery (%) | ||||
|---|---|---|---|---|---|---|---|---|
| Pb2+ | Co2+ | Cd2+ | Ni2+ | Cr3+ | ||||
a 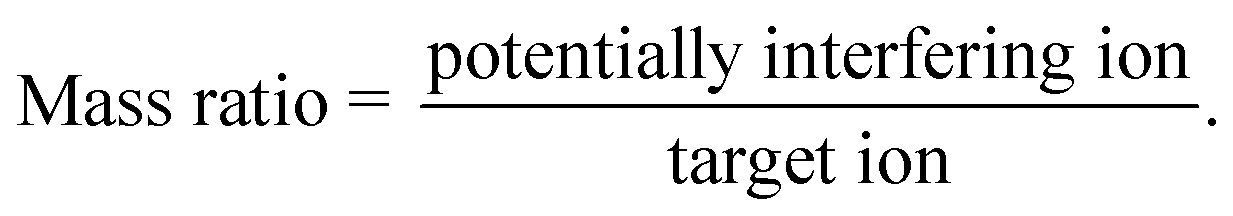 |
||||||||
| Li+ | 600 | LiNO3 | 12000 |
98.5 | 97.3 | 95.9 | 96.4 | 98.7 |
| Na+ | 600 | NaCl | 12000 |
96.6 | 98.2 | 97.1 | 95.3 | 98.0 |
| K+ | 600 | KCl | 12000 |
97.9 | 96.7 | 98.2 | 95.1 | 101.5 |
| Ag+ | 40 | AgNO3 | 800 | 96.1 | 98.4 | 97.3 | 98.6 | 99.2 |
| NH4+ | 500 | NH4NO3 | 10000 |
102.1 | 99.2 | 96.5 | 97.3 | 100.5 |
| Mg2+ | 55 | MgCl2·6H2O | 1100 | 97.5 | 98.1 | 96.4 | 95.2 | 99.1 |
| Ca2+ | 50 | CaCl2 | 1000 | 101.4 | 97.6 | 99.2 | 95.1 | 96.8 |
| Ba2+ | 47.5 | BaCl2 | 950 | 99.8 | 95.7 | 98.3 | 95.4 | 98.1 |
| Fe2+ | 42.5 | FeCl2·6H2O | 850 | 98.6 | 97.2 | 99.7 | 95.5 | 96.3 |
| Cu2+ | 2.25 | Cu(NO3)2·6H2O | 45 | 95.3 | 96.4 | 95.9 | 95.1 | 96.2 |
| Zn2+ | 2.4 | Zn(NO3)2·6H2O | 48 | 95.1 | 95.5 | 95.4 | 96.2 | 97.5 |
| Mn2+ | 45 | Mn(NO3)2·6H2O | 900 | 99.8 | 101.3 | 96.6 | 98.4 | 102.5 |
| Al3+ | 40 | Al(NO3)3·9H2O | 800 | 98.2 | 99.1 | 97.9 | 96.2 | 95.4 |
| F− | 600 | NaF | 12000 |
98.3 | 96.2 | 95.5 | 97.1 | 96.4 |
| Cl− | 600 | NaCl | 12000 |
99.4 | 96.1 | 97.9 | 98.0 | 102.6 |
| Br− | 500 | NaBr | 10000 |
98.1 | 99.7 | 96.3 | 98.8 | 98.3 |
| NO3− | 600 | NaNO3 | 12000 |
101.8 | 97.4 | 97.7 | 96.3 | 99.6 |
| CH3COO− | 250 | CH3COONa | 5000 | 98.7 | 95.1 | 95.4 | 98.3 | 96.8 |
| SO42− | 42.5 | Na2SO4 | 850 | 95.6 | 97.3 | 95.8 | 96.7 | 95.2 |
| CO32− | 45 | Na2CO3 | 900 | 96.3 | 95.9 | 95.4 | 98.3 | 96.8 |
| PO43− | 40 | Na3PO4 | 800 | 99.2 | 98.3 | 95.1 | 96.4 | 95.5 |
3.5. Analytical performance of method
Under the above-mentioned optimized conditions, calibration plots have a linear response in the range of 0.9–980 μg L−1 with the determination coefficient (r2) more than 0.992. Limits of detection (LODs) were calculated as three times the standard deviation of ten replicate runs of samples spiked with a low concentration of the analytes (10.0 μg L−1). LODs were in the range of 0.3 to 2.0 μg L−1 for the analytes. Intra- and inter-day precisions were determined at three spiked levels of the target ions (6.0, 40.0, and 70.0 μg L−1) with five analyses on the same day and over five different days. The results obtained showed good relative standard deviations (RSDs) ranging from 3.4 to 4.2% and 4.1 to 5.3% for the intra- and inter-day precisions, respectively (Table 4).| Ions | LODb (μg L−1) | LDRc (μg L−1) | Intra-day precision (%) | Inter-day precision (%) | EFd |
|---|---|---|---|---|---|
| a Experimental conditions: TA: MWCNTs, AA: 1.6 mg, CL: 0.07 mol L−1, pH: 6.4, NEC: 10, TE: HNO3, VE: 300 μL, and CE: 3.5 mol L−1.b n = 7.c Linear dynamic range.d n = 3. | |||||
| Pb2+ | 2.0 | 5.0–980 | 3.4 | 4.6 | 30 ± 1 |
| Cd2+ | 0.3 | 0.9–80 | 4.2 | 4.8 | 31 ± 1 |
| Ni2+ | 2.0 | 5.0–640 | 3.5 | 4.3 | 30 ± 1 |
| Cr3+ | 2.0 | 4.0–478 | 3.8 | 5.3 | 30 ± 1 |
| Co2+ | 2.0 | 4.0–497 | 3.9 | 4.1 | 29 ± 1 |
3.6. Application of SA-DM-SPE to analysis of real samples
The SA-DM-SPE method was applied for extraction of the Pb2+, Co2+, Ni2+, Cd2+, and Cr3+ ions in different biological fluid (saliva and urine), fruit juice (apple, pear, grape, and grapefruit), and water (tap and wastewater) samples prior to their determination using the MS-FAAS technique. For analysis of the samples, standard addition method was used, and the analytical results were tabulated in Table 5. As can be seen, satisfactory agreement obtained between the added and measured amounts of the metal ions indicates the capability of the method for determination of the interested ions in different samples. The method was validated by determining the certified reference materials (CRMs), CRMTMDW-500 and NIST SRM 1568a. The obtained results were in good agreement with the certified values in the CRMs (Table 1S†). It can be concluded that the proposed method is accurate and free from systematic errors.| Sample | Co2+ | Pb2+ | Ni2+ | Cd2+ | Cr3+ | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Added (μg L−1) | Found (found-real) (μg L−1) | RRa (%) | Added (μg L−1) | Found (found-real) (μg L−1) | RR (%) | Added (μg L−1) | Found (found-real) (μg L−1) | RR (%) | Added (μg L−1) | Found (found-real) (μg L−1) | RR (%) | Added (μg L−1) | Found (found-real) (μg L−1) | RR (%) | |
| a Relative recovery, n = 3.b Standard deviation.c Below detection limit. | |||||||||||||||
| Urine | 0.0 | 6.8 ± 0.32b | — | 0.0 | 31.7 ± 1.6 | — | 0.0 | 28.6 ± 1.3 | — | 0.0 | 10.2 ± 0.46 | — | 0.0 | 37.8 ± 1.9 | — |
| 10.0 | (9.7 ± 0.45) | 97 | 10.0 | (9.8 ± 0.44) | 98 | 10.0 | (10.1 ± 0.43) | 101 | 10.0 | (9.7 ± 0.42) | 97 | 10.0 | (10.1 ± 0.44) | 101 | |
| Saliva | 0.0 | BDLc | — | 0.0 | 6.6 ± 0.32 | — | 0.0 | 5.4 ± 0.25 | — | 0.0 | BDL | — | 0.0 | 7.1 ± 0.31 | — |
| 5.0 | (4.9 ± 0.23) | 98 | 10.0 | (9.7 ± 0.24) | 97 | 5.0 | (4.8 ± 0.21) | 96 | 5.0 | (4.7 ± 0.22) | 94 | 10.0 | (9.9 ± 0.44) | 99 | |
| Apple juice | 0.0 | 18.3 ± 0.92 | — | 0.0 | 520.8 ± 25.5 | — | 0.0 | 61.3 ± 2.9 | — | 0.0 | BDL | — | 0.0 | 38.6 ± 1.8 | — |
| 5.0 | (4.8 ± 0.24) | 96 | 50.0 | (50.5 ± 2.4) | 101 | 10.0 | (9.7 ± 0.47) | 97 | 5.0 | (4.8 ± 0.23) | 96 | 10.0 | (9.7 ± 0.45) | 97 | |
| Pear juice | 0.0 | 9.6 ± 0.46 | — | 0.0 | 223.5 ± 11.2 | — | 0.0 | 80.3 ± 4.0 | — | 0.0 | BDL | — | 0.0 | 22.3 ± 1.1 | — |
| 10.0 | (9.9 ± 0.43) | 99 | 50.0 | (48.5 ± 2.3) | 97 | 10.0 | (9.8 ± 0.46) | 98 | 5.0 | (4.8 ± 0.22) | 96 | 10.0 | (9.8 ± 0.14) | 98 | |
| Grape juice | 0.0 | BDL | — | 0.0 | 78.7 ± 3.8 | — | 0.0 | 69.4 ± 3.1 | — | 0.0 | 6.7 ± 0.31 | — | 0.0 | 28.9 ± 1.3 | — |
| 5.0 | (4.8 ± 0.22) | 96 | 50.0 | (51.0 ± 2.4) | 102 | 10.0 | (9.9 ± 0.46) | 99 | 10.0 | (9.5 ± 0.47) | 95 | 10.0 | (10.0 ± 0.49) | 100 | |
| Grapefruit juice | 0.0 | 17.8 ± 0.81 | — | 0.0 | 386.8 ± 18.6 | — | 0.0 | 94.5 ± 4.7 | — | 0.0 | 38.7 ± 1.9 | — | 0.0 | 17.6 ± 0.82 | — |
| 10.0 | (9.5 ± 0.44) | 95 | 50.0 | (49.2 ± 2.3) | 98 | 10.0 | (10.2 ± 0.47) | 102 | 10.0 | (9.8 ± 0.49) | 98 | 10.0 | (9.9 ± 0.48) | 99 | |
| Tap water (Semnan) | 0.0 | BDL | — | 0.0 | 32.3 ± 1.6 | — | 0.0 | 11.9 ± 0.58 | — | 0.0 | BDL | — | 0.0 | 93.5 ± 4.3 | — |
| 5.0 | (4.7 ± 0.22) | 94 | 10.0 | (9.5 ± 0.46) | 95 | 10.0 | (9.8 ± 0.47) | 98 | 5.0 | (4.8 ± 0.23) | 96 | 10.0 | (9.8 ± 0.47) | 98 | |
| Wastewater (Semnan) | 0.0 | 214.5 ± 10.5 | — | 0.0 | 334.6 ± 15.1 | — | 0.0 | 146.7 ± 7.2 | — | 0.0 | 173.9 ± 7.8 | — | 0.0 | 289.4 ± 13.9 | — |
| 50.0 | (47.5 ± 2.4) | 95 | 50.0 | (48.5 ± 0.45) | 97 | 50.0 | (49.5 ± 2.4) | 99 | 50.0 | (48.5 ± 2.3) | 97 | 50.0 | (48.5 ± 2.2) | 97 | |
3.7. Comparison of SA-DM-SPE with other reported methods
A comparison between the characteristics of the proposed method and some of the reported methods for the extraction and determination of the target ions in different real samples is shown in Table 6. In comparison with other extraction methods, the SA-DM-SPE method has some advantages including:| Method | Matrix | Metal ions | LOD | Recovery | jPreconcentration factor (Volume of sample) | Consumptive index | Final volume of eluent | Amount of adsorbent | Extraction time (adsorption and desorption steps) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|
| a Adsorbent: multi-walled carbon nanotubes.b Adsorbent: nano-alumina coated with sodium dodecyl sulfate-1-(2-pyridylazo)-2-naphthol.c Adsorbent: gold nanoparticle loaded in activated carbon and modified by bis(4-methoxy salicylaldehyde)-1,2-phenylenediamine.d Adsorbent: multiwalled carbon nanotubes chemically functionalized with 2-((3-silylpropylimino) methyl) phenol.e Adsorbent: guanidin functionalized SBA-15.f Adsorbent: magnetic metal organic frame work immobilized with Fe3O4–dithizone.g Adsorbent: chemically functionalized multi-walled carbon nanotubes with 3-hydroxy-4-((3-silylpropylimino) methyl) phenol.h Adsorbent: 1-(2-pyridylazo)-2-naphthol impregnated activated carbon cloth.i Adsorbent: multi-walled carbon nanotubes.j Since reported recoveries are frequently near to 100%, it supposed that the preconcentration and enrichment factors are equal, unless the values had been separately mentioned in the papers. | ||||||||||
| Solid-phase extractiona/FAAS | Food and real water samples | Cu2+, Cd2+, Pb2+, Zn2+, Ni2+ and Co2+ | 0.3–0.6 μg L−1 | 95.0–98.0% | 80 (400 mL) | ∼5.0 | 5 mL | 300 mg | ∼12 min | 36 |
| Solid-phase extractiona/FAAS | Herbal plants, food and real water samples | Fe2+, Cu2+, Mn2+ and Pb2+ | 3.5–8.0 μg L−1 | 95.2–106.0% | 20 (100 mL) | ∼5.0 | 5 mL | 100 mg | ∼35 min | 37 |
| Solid-phase extractionb/FAAS | Food and real water samples | Cd2+ and Pb2+ | 0.15 and 0.17 μg L−1 | 97.3–105.4% | 250 (500 mL) | ∼2.0 | 2 mL | 50 mg | ∼45 min | 38 |
| Solid-phase extractionc/FAAS | Food samples | Co2+, Cu2+, Ni2+, Fe2+, Pb2+ and Zn2+ | 1.4–2.6 μg L−1 | 94.0–106.0% | 267 (1600 mL) | ∼8.0 | 6 mL | 300 mg | ∼84 min | 39 |
| Solid-phase extractiond/FAAS | Fruit and vegetable samples | Cu2+, Pb2+, Fe2+, Ni2+, and Zn2+ | 1.0–2.6 μg L−1 | 94.4–104.0% | 100 (600 mL) | ∼6.0 | 6 mL | 150 mg | ∼100 min | 40 |
| Dispersive solid-phase extractione/FAAS | Food and water samples | Pb2+, Cu2+, Zn2+ and Cd2+ | 0.2–4.5 μg L−1 | 98.0–100.1% | 100 (2500 mL) | ∼25.0 | 25 mL | 10 mg | ∼20 min | 41 |
| Dispersive solid-phase extractionf/FAAS | Fish, sediment, soil, and water samples | Cd2+, Pb2+, Ni2+, and Zn2+ | 0.12–1.2 μg L−1 | 90.0–104.0% | 128 (1000 mL) | ∼8.0 | 7.8 mL | 25 mg | ∼32 min | 42 |
| Dispersive solid-phase extractiong/FAAS | Fruit and vegetable samples | Cu2+, Ni2+, Zn2+, Pb2+, Co2+ and Fe3+ | 1.0–2.6 μg L−1 | 96.0–106.0% | 267 (1600 mL) | ∼8.0 | 6 mL | 300 mg | ∼100 min | 43 |
| Solid-phase extractionh/FAAS | Soil and environmental water samples | Cd2+, Pb2+ and Ni2+ | 0.1–2.8 μg L−1 | 95.0–104.0% | 100 (1000 mL) | ∼10.0 | 10 mL | Not reported | ∼28 min | 44 |
| Surfactant mediated magnetic solid-phase extraction/FAAS | Water and soil samples | Cd2+ and Pb2+ | 0.15 and 0.74 μg L−1 | 98.4–100.0% | 25 (10 mL) | ∼0.40 | 400 μL | 50 mg | ∼20 min | 45 |
| Syringe-assisted dispersive micro solid-phase extractioni/FAAS | Water, fruit juice and biological fluid samples | Pb2+, Cd2+, Co2+, Ni2+ and Cr3+ | 0.3 to 2.0 μg L−1 | 94.0–102.0% | 33 (10 mL) | ∼0.33 | 300 μL | 1.6 mg | ∼1 min | This research |
(i) It is more environmental friendly, due to consumption of low amount of eluent.
(ii) It is simpler and faster, performing in fewer steps.
(iii) The analytical merits are comparable to other extraction methods.
(iv) A small amount of adsorbent is required to achieve acceptable recoveries.
(v) Higher enrichment factors are achieved, when equal volumes of the samples are considered. This provides comparable or even better LODs than other methods.
The superiority of the SA-DM-SPE can be demonstrated with a useful term, named consumptive index (CI), which is defined as:
 | (9) |
4. Conclusion
In this work, an optimized syringe-assisted dispersive micro solid phase extraction method was developed for the extraction of some potentially toxic metal ions, as model analytes, from different real samples, prior to their determination using a micro-sampling flame atomic absorption spectrometry technique. The method exhibited the following merits:(i) Adsorption of the chelated ions onto the adsorbent (MWCNTs) was very fast, and was performed with the aid of a single syringe, which avoided the requirement to accelerate mass transfer assistants such as sonication and vortex.
(ii) A very small amount of adsorbent (1.6 mg of MWCNTs) was required to achieve acceptable recoveries of the target ions.
(iii) The method was performed with no need for centrifugation, which is time-consuming and is essentially an off-line step. It opens up a new horizon to the automation of the dispersive micro solid phase extraction method.
(iv) The application of experimental design also provided a large amount of information concerning the factor-response behavior of the method with a minimum number of experiments.
(v) The results obtained shows that the SA-DM-SPE method offers low limits of detection and consumptive indices, acceptable repeatabilities, wide linear dynamic ranges, and good recoveries.
Overall, the optimized SA-DM-SPE method offers an attractive alternative for the extraction of potentially toxic metals from real samples, providing several advantages including fewer steps, faster sample throughput, and ease of performance (using single devices) compared to the commonly used DM-SPE methods. These significant features are of key interest for the routine trace metal laboratory analysis, which could be extended to the analysis of other inorganic and organic compounds.
The authors have declared no conflict of interest.
Acknowledgements
The authors would like to thank the Semnan University Research Council for the financial support of this work.References
- S. Granero and J. Domingo, Environ. Int., 2002, 28, 159–164 CrossRef CAS.
- S. Huang, Q. Liao, M. Hua, X. Wu, K. Bi, C. Yan, B. Chen and X. Zhang, Chemosphere, 2007, 67, 2148–2155 CrossRef CAS PubMed.
- J. A. Baig, T. G. Kazi and L. Elci, Separ. Sci. Technol., 2012, 47, 1044–1054 CrossRef CAS.
- M. Ghaedi, B. Karami, S. Ehsani, F. Marahel and M. Soylak, J. Hazard. Mater., 2009, 172, 802–808 CrossRef CAS PubMed.
- M. Rajabi, B. Barfi, A. Asghari, F. Najafi and R. Aran, Food Anal. Method, 2014, 1–10 Search PubMed.
- M. Rajabi, S. Asemipour, B. Barfi, M. R. Jamali and M. Behzad, J. Mol. Liq., 2014, 194, 166–171 CrossRef CAS PubMed.
- A. Asghari and B. Mohammadi, J. Ind. Eng. Chem., 2014, 20, 824–829 CrossRef CAS PubMed.
- B. L. Batista, J. L. Rodrigues, J. A. Nunes, L. Tormen, A. J. Curtius and F. Barbosa Jr, Talanta, 2008, 76, 575–579 CrossRef CAS PubMed.
- M. Rajabi, B. Mohammadi, A. Asghari, B. Barfi and M. Behzad, J. Ind. Eng. Chem., 2014, 20, 3737–3743 CrossRef CAS PubMed.
- B. Barfi, M. Rajabi, M. M. Zadeh, M. Ghaedi, M. Salavati-Niasari and R. Sahraei, Microchim. Acta, 2015, 182, 1187–1196 CrossRef CAS.
- T. Wai, H. Darus and N. Mohamed, Talanta, 1996, 43, 1539–1544 CrossRef CAS.
- J. A. Baig, A. Hol, A. Akdogan, A. A. Kartal, U. Divrikli, T. G. Kazi and L. Elci, J. Anal. At. Spectrom., 2012, 27, 1509–1517 RSC.
- A. Asghari, B. Barfi, A. Barfi, I. Saeidi, F. Ghollasi Moud, M. Peyrovi and A. Beig Babaei, Acta Chromatogr., 2014, 26, 157–175 CrossRef CAS.
- S. Tang and H. K. Lee, Anal. Chem., 2013, 85, 7426–7433 CrossRef CAS PubMed.
- M. R. Jamali, A. Firouzjah and R. Rahnama, Talanta, 2013, 116, 454–459 CrossRef CAS PubMed.
- M. Asensio-Ramos, G. D'Orazio, J. Hernandez-Borges, A. Rocco and S. Fanali, Anal. Bioanal. Chem., 2011, 400, 1113–1123 CrossRef CAS PubMed.
- B. Barfi, M. R. Hadjmohammadi and M. R. Kasaai, Monatsh. Fur Chem., 2009, 140, 1143–1148 CrossRef CAS PubMed.
- B. Barfi, A. Asghari, M. Rajabi, A. Barfi and I. Saeidi, J. Chromatogr. A, 2013, 1311, 30–40 CrossRef CAS PubMed.
- K. Hylton and S. Mitra, J. Chromatogr. A, 2007, 1152, 199–214 CrossRef CAS PubMed.
- F. David and P. Sandra, J. Chromatogr. A, 2007, 1152, 54–69 CrossRef CAS PubMed.
- J. Feng, H. Qiu, X. Liu and S. Jiang, Trac-Trend Anal. Chem., 2013, 46, 44–58 CrossRef CAS PubMed.
- M. Rajabi, S. Haji-Esfandiari, B. Barfi and H. Ghanbari, Anal. Bioanal. Chem., 2014, 406, 4501–4512 CrossRef CAS PubMed.
- P. Berton, E. M. Martinis, L. D. Martinez and R. G. Wuilloud, Anal. Chim. Acta, 2012, 713, 56–62 CrossRef CAS PubMed.
- A. Asghari, H. Fazl-Karimi, B. Barfi, M. Rajabi and A. Daneshfar, Hum Exp. Toxicol., 2013, 33, 863–872 Search PubMed.
- M. Rajabi, H. Ghanbari, B. Barfi, A. Asghari and S. Haji-Esfandiari, Food Res. Int., 2014, 62, 761–770 CrossRef CAS PubMed.
- F. Galán-Cano, R. Lucena, S. Cárdenas and M. Valcárcel, Anal. Method, 2011, 3, 991–995 RSC.
- G. Lasarte-Aragonés, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2011, 1218, 9128–9134 CrossRef PubMed.
- G. Lasarte-Aragonés, R. Lucena, S. Cárdenas and M. Valcárcel, Anal. Bioanal. Chem., 2013, 405, 3269–3277 CrossRef PubMed.
- K. M. Giannoulis, G. Z. Tsogas, D. L. Giokas and A. G. Vlessidis, Talanta, 2012, 99, 62–68 CrossRef CAS PubMed.
- M. García-Valverde, R. Lucena, F. Galán-Cano, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2014, 1343, 26–32 CrossRef PubMed.
- E. M. Reyes-Gallardo, G. Lasarte-Aragonés, R. Lucena, S. Cárdenas and M. Valcárcel, J. Chromatogr. A, 2013, 1271, 50–55 CrossRef CAS PubMed.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. Kocot, B. Zawisza, E. Marguí, I. Queralt, M. Hidalgo and R. Sitko, J. Anal. At. Spectrom., 2013, 28, 736–742 RSC.
- S. Özkar, D. Ülkü, L. T. Yıldırım, N. Biricik and B. Gümgüm, J. Mol. Struct., 2004, 688, 207–211 CrossRef PubMed.
- P. Olmedo, A. Pla, A. F. Hernández, O. López-Guarnido, L. Rodrigo and F. Gil, Anal. Chim. Acta, 2010, 659, 60–67 CrossRef CAS PubMed.
- M. Tuzen, K. O. Saygi and M. Soylak, J. Hazard. Mater., 2008, 152, 632–639 CrossRef CAS PubMed.
- S. G. Ozcan, N. Satiroglu and M. Soylak, Food Chem. Toxicol., 2010, 48, 2401–2406 CrossRef CAS PubMed.
- M. Ezoddin, F. Shemirani, K. Abdi, M. K. Saghezchi and M. Jamali, J. Hazard. Mater., 2010, 178, 900–905 CrossRef CAS PubMed.
- G. Karimipour, M. Ghaedi, R. Sahraei, A. Daneshfar and M. N. Biyareh, Biol. Trace Elem. Res., 2012, 145, 109–117 CrossRef CAS PubMed.
- M. Ghaedi, M. Montazerozohori, M. N. Biyareh, K. Mortazavi and M. Soylak, Int. J. Environ. Anal. Chem., 2013, 93, 528–542 CrossRef CAS.
- L. Hajiaghababaei, T. Tajmiri, A. Badiei, M. R. Ganjali, Y. Khaniani and G. M. Ziarani, Food Chem., 2013, 141, 1916–1922 CrossRef CAS PubMed.
- M. Taghizadeh, A. A. Asgharinezhad, M. Pooladi, M. Barzin, A. Abbaszadeh and A. Tadjarodi, Microchim. Acta, 2013, 180, 1073–1084 CrossRef CAS.
- M. Ghaedi, M. Montazerozohori, N. Rahimi and M. N. Biysreh, J. Ind. Eng. Chem., 2013, 19, 1477–1482 CrossRef CAS PubMed.
- Z. A. Alothman, E. Yilmaz, M. Habila and M. Soylak, Ecotox. Environ. Safe., 2015, 112, 74–79 CrossRef CAS PubMed.
- N. Jalbani and M. Soylak, Ecotox. Environ. Safe., 2014, 102, 174–178 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03537f |
| This journal is © The Royal Society of Chemistry 2015 |