
The hydrolysis of phenyl trifluoroacetate in AOT/n-heptane RMs as a sensor of the encapsulated water structure†
Abstract
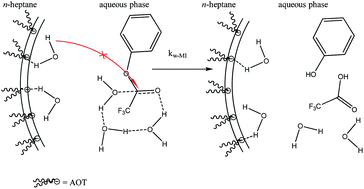
A study was carried out on the hydrolysis of phenyl trifluoroacetate (PTFA) in AOT/n-heptane/water reverse micelles. The results obtained have been interpreted by taking into account the distribution of the PTFA using a kinetic model based on the pseudophase concept and, it is demonstrated that the reaction takes place in the water pool of the reverse micelles. Moreover, it is established that the water molecules involved in the hydration of the interface do not react with PTFA or they do it at much slower rate compared with the water molecules that form the pool. The value of the second order rate constant in the water pool, kw–MI, is of the same order of magnitude as that in pure water but is greater (500 times) than those obtained in water–acetonitrile mixtures with low water content. We performed kinetic solvent isotopic effect (KSIE) and proton inventory experiments (PI) and we concluded that the water is acting as a general-base catalyst and the water sequestered in the water pool is pre-organized favoring the activation process, in contrast to what happened in water–acetonitrile mixtures.
 Please wait while we load your content...
Please wait while we load your content...

