
Single-step label-free hepatitis B virus detection by a piezoelectric biosensor†
Nicoletta Giamblanco*a,
Sabrina Conoci*b,
Dario Russoc and
Giovanni Marlettaa
aLaboratory for Molecular Surfaces and Nanotechnology (LAMSUN), Department of Chemical Sciences, University of Catania and CSGI, 95125 Catania, Italy. E-mail: n.giamblanco@unict.it
bSTMicroelectronics, Stradale Primosole 50, 95121 Catania, Italy. E-mail: sabrina.conoci@st.com
cClonit Srl, Via Bernardo Quaranta 57, 20139 Milano MI, Italy
First published on 14th April 2015
Abstract
In this paper we describe a single step and label free method to selectively detect the HVB genome based on hybridization with simple linear ssDNA probes immobilized on the Au surfaces of a QCM resonator. It has been proved that selective sensing ability is obtained for a large DNA target, constituted by an HBV clone of 7 kbps through a proper optimization of the probe density. Actually, with a probe density of ∼4.0 × 1012 molecules per cm2 we were able to detect fmol cm−2 of HBV target, without using any amplification steps or labelling method. The results presented herein pave the way to the development of an easy to use and portable PoC sensor device for direct and fast HBV detection.
Introduction
The development of nucleic acid biosensors has received increasing attention over the last decade, due to the importance of gene analysis1 in clinical diagnostics and forensic studies.2 In this context, most of the consolidated nucleic acid detection methods are time consuming and expensive. Consequently, there is a high demand for accurate biosensors with rapid detection systems in a portable Point-of-Care (PoC) format.Particularly, for hepatitis B virus (HBV) detection, there is huge request to detect and monitor at early stage of infection since it is one of the major health problems worldwide leading to chronic hepatitis, cirrhosis and primarily liver cancer.3,4
The hepatitis B virus (HBV) is a mostly double-stranded DNA virus in the Hepadnaviridae family. The HBV virion genome is circular and approximately 3.2 kb in size and consists of DNA that is mostly double stranded. It has compact organization, with four overlapping reading frames running in one direction and no noncoding regions.5
Currently, the most commonly used clinical diagnostic methods for HBV detection are based on immunoassay and polymerase chain reaction (PCR). Immunoassays present a good selectivity and achieve 100% accuracy, but they do not provide quantitative results and the detection is limited by serological response.6 On the other hands, PCR, that is traditionally the clinical standard technique to quantitatively identify HBV infection, requires complex and high cost instrumentation.7
DNA biosensors based on nucleic acids hybridization are currently under intense investigation owing to their increasing importance in the diagnosis of diseases, with low cost and low power requirements.8–10
Among the methods developed for hybridization analysis, the most attractive ones for portable PoC devices are those based on electronic detection since they offer the advantages to be easily integrated with microelectronics in miniaturized chip-based format.11,12 Recently, increasing attention has been paid to the development of quartz crystal microbalance (QCM) biosensors due to their many merits such as compact size, high mass sensitivity, easiness to be properly functionalised in view of good specificity, low cost, label-free detection and rapid response.13–16 Indeed, QCM detection methods require device architectures implying to immobilize specific DNA probes on Au surfaces. However, such methodologies require the control of the accessibility, in order to enable the full accessibility of the detection sites on the probe. In fact, the immobilized probe molecules may suffer a significant reduction of the appropriate exposure of the recognizing sites, when compared to the efficiency of probe-target recognition in solution. Despite extensive investigations at this regard17–23 only a limited number of studies have been addressed to the detection of large DNA target without amplification.24,25
Accordingly, the performances of QCM-based biosensor devices (i.e. specificity, sensitivity and reproducibility) are supposed to strictly depend on the control of both homogeneity and structural features of the immobilized DNA probes, respectively involving packing density and molecular conformation.26–29 One of the approach to improve biosensor ability and overcome the limitations in the case of large DNA target recognition, is therefore to design devices based on complex nanoarchitectures of DNA probes30,31 involving multistep surface chemistry.32 Again, this would represent a serious drawback for an industrial scale-up of the process.
In order to face the problems described above, in this paper we describe a single step and label free method to perform gene analysis, showing its application, as a model system, to the selective detection of HVB genome based on hybridization with simple linear ssDNA probes immobilized on Au surfaces of QCM resonator. In particular, it is shown that fast and selective sensing ability is obtained for large DNA target, constituted by HBV clone of 7 kbps through a proper optimization of the probe density. The results presented herein are very promising for the development of an easy-to-use, portable PoC sensors device for direct and fast HBV detection.
Experimental
2.1 Materials and methods
Thiolated oligonucleotides probes HBS-F (henceforth indicated as P1) and HBS-R (henceforth indicated as P2) (20, 21 nts) were purchased from Eurofins MWG Operon and were prepared with concentration range 0.08–38.8 μM in ultrapure water (DNase and RNase-free, Gibco). The ultrapure water osmolality is certified to be 1.0 mOsm kg−1 with pH 6.0.Stock solutions of hepatitis B virus (HBV) clone complete genome (T-clone) (ref. 05960467, consisting in HBV genome 3.2 kbps and a plasmid PBR322 vector 3.8 kbps) in TE (Tris 10 mM, EDTA 1 mM, pH = 8), R-clone (ref. RT 50, consisting of Factor V of Leiden of 6062 kbps) in TE (Tris 10 mM, EDTA 1 mM, pH = 8) were provided by Clonit. These were diluted at concentration of 370 pg in ultrapure water (pH = 8.1 ± 0.2). Both these solutions (T-clone and R-clone) were denatured at 95 degrees for 5′ and immediately immersed in ice bath before use. The recognition site on hepatitis B-virus for ssDNA probe sequences P1 and P2 are located at the core protein coded for by gene C (HBcAg)33 at nucleotide position 1 (P2) and 181 (P1), respectively (Table 1).
| Name | Sequences (5′ to 3′) | Size | MW (g mol−1) |
|---|---|---|---|
| P1 | HBS-F CACATCAGGATTCCTAGGAGG | 21 nts | 6570 |
| P2 | HBS-R GGTGAGTGATTGGAGGTTGG | 20 nts | 6519 |
| T-clone | HBV | 7000 bps | 4.6 × 106 |
| R-clone | Factor V of Leiden | 6062 bps | 4.1 × 106 |
2.2 Instrumentation
(a) Probe immobilization study: it started by flowing ultrapure water solutions (H2O), followed by thiolated ssDNA probes (P1 and P2) solutions at various concentrations (38.8 μM, 7.7 μM, 0.8 μM and 0.08 μM) with nuclease free water and monitoring the related frequency and dissipation changes. After that, rinsing steps are carried out to remove the excess of probe not linked to the surface.
(b) Target recognition: it started by flowing at first solution of R-clone, at a concentration of 370 pg, followed by a rinsing step, and by the subsequent injection of the specific genome target (T-clone) with a final rinsing step. Ultrapure water was used during the probe immobilization and target hybridization steps.
All measurements were performed at temperature of 25 °C. At the frequency overtones, 15 MHz, 25 MHz, 35 MHz, 45 MHz, 55 MHz and 65 MHz, the Δf shifts and ΔD were obtained with an accuracy of ±0.2 Hz and ±0.2 × 10−6 respectively. Frequency (Δf) and dissipation (ΔD) shifts were measured with respect to a baseline obtained in ultrapure water.
Results and discussion
3.1 Probe anchoring study
Fig. 1 reports the measured real time acoustic QCM-D curves in a concentration range of 0.8–38.8 μM for the probe P1 (Fig. 1a) and probe P2 (Fig. 1b). In this range, small but detectable amounts of adsorbed probes are found; while below the concentration of 0.8 μM (i.e., 0.08 μM) no significant adsorption has been found.
 | ||
| Fig. 1 Real time acoustic QCM-D curves (15 MHz). Frequency changes due to the adsorption of thiolated ssDNA probe P1 (a) and probe P2 (b) on gold are reported for solution concentrations of 38.8 μM (triangles), 7.7 μM (square), 0.8 μM (circle). Each sample addition is followed by the rinsing step indicated with arrows (H2O). | ||
According to Fig. 1, three kinetic steps can be identified: (i) probe adsorption (Δf < 0) during the surface exposure at the probe solutions; (ii) small probe desorption (Δf > 0) during the rinsing step; (iii) a further step, after rinsing, where the frequency decrease again (Δf < 0). According to literature,34 we propose that the ssDNA molecules are anchored to the gold, via the thiol group, in a standing-up position. The further frequency decreasing after the rinsing step (iii) can be attributed to the adsorption of a water shell around the DNA molecules, enabling the ssDNA molecules to extend farther out into the water according to literature.34
Table 2 reports the averaged values measured for frequency and dissipation changes upon P1 and P2 probe adsorption. Since the measured energy dissipation ΔD is low, the Sauerbrey frequency–mass relationship can be used to calculate the amount of anchored probe.35 According to Sauerbrey, the relationship between frequency change (Δf) and mass adsorbed (Δm) is given by Δm = −ΔfCn, where n is the harmonic number and C is a experimental constant characteristic of the employed sensor and equal to ∼−17.7 ng cm−2 Hz−1 for a 5 MHz crystal. The measured averaged Δf values for the three solution concentrations of probe P1, after the first hour of adsorption, were −7.5 ± 0.3 Hz (38.8 μM), −5.0 ± 0.4 Hz (7.7 μM) and −2.8 ± 0.2 Hz (0.8 μM), respectively corresponding to about 132.75 ± 5.3 ng cm−2, 88.5 ± 7.8 ng cm−2 and 49.6 ± 3.5 ng cm−2 of absorbed mass. According to that, the respective ssDNA probe surface densities can be estimated to be 1.2 ± 0.6 × 1013 molecule per cm2 (from 38.8 μM solution), 8.2 ± 0.5 × 1012 molecule per cm2 (from 7.7 μM) and 4.6 ± 0.3 × 1012 molecule per cm2 (from 0.8 μM) respectively. Similar results for the adsorption process with slightly lower surface density values were measured for the P2 ssDNA probe (Fig. 1b and Table 2b). The obtained surface density values are in good agreement with the literature for similar anchoring studies.18
| (a) P1 probe | ||||
|---|---|---|---|---|
| [P1] conc. (μM) | ΔD (10−6) | Δf (Hz) | Number of molecules per cm2 (1012) | d (nm) |
| 38.8 | 2.5 ± 0.3 | −7.5 ± 0.3 | 12.0 ± 0.6 | 2.9 ± 0.2 |
| 7.7 | 1.9 ± 0.2 | −5.0 ± 0.4 | 8.2 ± 0.5 | 3.5 ± 0.1 |
| 0.8 | 0.6 ± 0.2 | −2.8 ± 0.2 | 4.6 ± 0.3 | 4.7 ± 0.1 |
| (b) P2 probe | ||||
|---|---|---|---|---|
| [P2] conc. (μM) | ΔD (10−6) | Δf (Hz) | Number of molecules per cm2 (1012) | d (nm) |
| 38.8 | 2.8 ± 0.3 | −6.2 ± 0.3 | 10.0 ± 0.6 | 3.2 ± 0.2 |
| 7.7 | 1.5 ± 0.2 | −4.0 ± 0.4 | 6.4 ± 0.5 | 3.9 ± 0.1 |
| 0.8 | 0.5 ± 0.2 | −2.3 ± 0.2 | 3.7 ± 0.3 | 5.2 ± 0.1 |
 | ||
| Fig. 2 Surface coverage (N molecules per cm2) versus solution concentration (C) of thiolated ssDNA probe (P1) after 1 hour. The correlation coefficient is R2 = 0.94. | ||
The equilibrium-binding constant of the ssDNA probes on gold (Ke) can be calculated by eqn (1),
| (dΓ(t))/dt = KeC(1 − (Γ(t))/Γ_max) | (1) |
The fitting of the experimental data by eqn (1), yields the following values for P1 and P2 equilibrium constants: Ke(P1) = 6.0 ± 1.0 × 105 M−1 and Ke(P2) = 4.0 ± 0.5 × 105 M−1.
The knowledge of the Ke values, hence, allows the derivation of the free energies of adsorption of thiolated ssDNA probes P1 and P2 on gold, according to the well known relation ΔG = −RT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) lnKe, where Ke = kads/kdes. In particular, from the values of Ke above reported, we obtained ΔGads(P1) = −42.0 ± 3.6 kJ mol−1 (−10.3 ± 0.9 kcal mol−1) and ΔGads(P2) = −43.0 ± 3.0 kJ mol−1 (−10.5 ± 0.7 kcal mol−1). These values are in close agreement with those expected for gold/thiol bond formation (−40 kJ mol−1).36 This finding supports the fact that the adsorption process essentially involves covalently bonded species, with a negligible role, if any, of non-specific interactions (i.e., van der Waals, image charge, etc.). Moreover, it has been recently proved that the Au–S bond formation (isolated molecules) is very fast (8 s)37 indicating that in our case, after 20 minutes (flowing time of probe surface exposure), all the thiolated probes are covalently bound.
lnKe, where Ke = kads/kdes. In particular, from the values of Ke above reported, we obtained ΔGads(P1) = −42.0 ± 3.6 kJ mol−1 (−10.3 ± 0.9 kcal mol−1) and ΔGads(P2) = −43.0 ± 3.0 kJ mol−1 (−10.5 ± 0.7 kcal mol−1). These values are in close agreement with those expected for gold/thiol bond formation (−40 kJ mol−1).36 This finding supports the fact that the adsorption process essentially involves covalently bonded species, with a negligible role, if any, of non-specific interactions (i.e., van der Waals, image charge, etc.). Moreover, it has been recently proved that the Au–S bond formation (isolated molecules) is very fast (8 s)37 indicating that in our case, after 20 minutes (flowing time of probe surface exposure), all the thiolated probes are covalently bound.
The data reported in Fig. 2 indicate that adsorption is roughly ranging between ∼3.0 × 1012 and 1.2 × 1013 molecules per cm2, reaching a plateau value thereafter, in agreement with similar results reported in literature.38,39
Also, the possible contribution of conformational and entropic forces, i.e., steric hindrance factors, to the distribution of the ssDNA strands along the gold surface have been analysed. In particular, we have compared the size of the free thiolated ssDNA molecules in solution with the estimated average distance among neighbouring chemisorbed molecules on gold, for the various measured surface densities. The contour lengths of fully stretched ssDNA strands is simply L = bN, where N is the number of bases in the strand and b ≅ 0.7 nm is the distance between adjacent phosphorous atoms.40 In our case L(P1) = 14.7 nm for N = 21 nts (P1 strands), and L(P2) = 14.0 nm for N = 20 nts (P2 strands). On the other hand, the real size of the thiolated ssDNA P1 strands can be described by means of the persistence length (lp), which, for the studied ssDNA strands is about 6.7 nm at the low ionic strength solutions here employed (i.e., ultrapure water).41 In this framework, the ssDNA molecules, under the condition lp < L, can be represented as semiflexible coils. In a simple bidimensional model, consisting of hexagonal close-packed arrangement,42 a distribution of weakly stretched ssDNA brushes,43 extending towards the solution, is therefore expected, according to well-established literature.44
3.2 Target recognition
Fig. 3 shows the measured real time hybridization QCM-D curves for the three surface densities cases of P1 probe: 1.2 × 1013 molecules per cm2, 8.2 × 1012 molecules per cm2 and 4.6 × 1012 molecules per cm2. It can be noticed that the most efficient hybridization is found for the probe density surfaces of 4.6 × 1012 molecules per cm2 (the lowest here explored) while the higher surface density samples show an almost negligible hybridization capability. In particular, Fig. 3a and b show that the surfaces with highest P1 probe density (1.2 × 1013 molecules per cm2 and 8.2 × 1012 molecules per cm2) produces a small and completely reversible adsorption for both the T- and R-clones, while for the low density surfaces (i.e., about 4.6 × 1012 molecules per cm2, Fig. 3c) a strongly selective and irreversible hybridization is found for T-clones, as shown by Fig. 3d, while the non-specific R-clones shows a negligible adsorption. In other words, the 4.6 × 1012 molecules per cm2 surface density samples show a dramatic increase in selective adsorption of T clone. Moreover, the low density surfaces retained about 45.0 ng cm−2 of T-clone mass (see Table 3), roughly corresponding to 1.2 × 1010 molecules per cm2 and, in turn, to 0.02 pmol cm−2, accordingly, the 4.6 × 1012 molecules per cm2 density surfaces may, in principle, reveal femtomolar concentrations.
 | ||
| Fig. 3 QCM-D kinetic curves of non-specific R-clones onto the ssDNA probe (P1), followed by washing with H2O and then flowed with specific T-clones and a final rinsing step. The ssDNA P1 probe density are (a) 1.2 × 1013 molecules per cm2; (b) 8.2 × 1012 molecules per cm2; (c) 4.6 × 1012 molecules per cm2. Panel (d) reports the clone to probe ratios for R/P1 and T/P1 for all the investigated ssDNA surface densities. | ||
| Probe P1 | R-clone | T-clone | ||
|---|---|---|---|---|
| N molecules per cm2 1012 | Δf (Hz) | Δm (ng cm−2) | Δf (Hz) | Δm (ng cm−2) |
| 12.0 ± 0.6 | −0.5 ± 0.2 | 8.8 ± 3.5 | −0.3 ± 0.2 | 5.3 ± 3.5 |
| 8.2 ± 0.5 | −0.5 ± 0.2 | 8.8 ± 3.5 | −0.3 ± 0.2 | 5.3 ± 3.5 |
| 4.6 ± 0.3 | −0.4 ± 0.2 | 7.1 ± 3.5 | −2.5 ± 0.3 | 45 ± 5.3 |
As far as the P2-is concerned, the best surface probe density T-clone recognition resulted the same than for P1 ssDNA species, i.e., about 4.0 × 1012 molecules per cm2, yielding a genome T-clone retained mass of 42 ng cm−2, very close to the one measured for P1 (Fig. SI4†).
It should be stressed that in this case non-specific R-clones were not adsorbed at all, indicating that neither recognition nor non-specific adsorption events occurred (see Fig. SI4†).
These findings highlight that the recognition of HBV genome is possible by a single step without requiring any amplification procedure.
 | ||
| Fig. 4 Height AFM images of gold surfaces functionalized with thiolated ssDNA from 7.7 μM (a) and 0.8 μM (c) solution; AFM images of the thiolated surfaces exposed to T-clone (b) and (d). | ||
The AFM results found a nice confirmation by comparing them to the data of surface densities for P1 and retained density of T-clone measured by QCM-D. Indeed, by calculating the hybridization HBV:P1 probes ratio, i.e., the retained 1.2 × 1010 molecules per cm2 HBV and the 4.6 × 1012 molecules per cm2 P1 probe density, a value of about 400:1 is obtained as well.
 | (2) |
| σT(t) = σeqT(1 − exp(kefft)) | (3) |
Fig. 5 reports the kinetic curves of mass change (ng cm−2) vs. t (s) for the two processes: (i) target hybridization (Δm > 0) during the surface exposure at the target solution; (ii) target excess desorption (Δm < 0) during the rinsing step.
 | ||
| Fig. 5 Mass uptake curves versus time (t) measured by means of QCM-D technique for specific genomic T-clone onto P1-functionalized gold sensors at various probe densities including 1.2 × 1013 molecules per cm2 (a), 8.2 × 1012 (b) and 4.6 × 1012 (c) molecules per cm2. | ||
All the curves of the hybridized target mass vs. time can be fitted by eqn (3) with Δmmax and keff are the fitting parameters.
In particular, since τ = 1/keff = (kaCT + kd)−1 and CT is the same (370 pg) for the three probe densities, τa ∼ (kaCT)−1 and τd ∼ kd−1.
Accordingly, Table 4 reports the τa values obtained by the fitting respectively for target hybridization and target excess desorption τd, at the three different surface densities of P1 probe.
| Probe P1 | T-clone | |
|---|---|---|
| N molecules per cm2 1012 | τa (s) | τd (s) |
| 12.0 ± 0.6 | 1601 ± 100 | 100 ± 20 |
| 8.2 ± 0.5 | 1002 ± 120 | 80 ± 10 |
| 4.6 ± 0.3 | 217 ± 20 | 65 ± 8.0 |
It can be seen, again, that the values of τa ∼ 210 s are obtained for the low probe density surfaces, suggesting that for these surfaces the P1 probe molecules are more accessible than at higher probe density, prompting the observed faster and efficient recognition of HBV-clone molecules (T-clones). Moreover, the high τd ∼ 1/kd values are almost comparable to kaCT, indicating that the surface concentration of bound targets (σT) will be less than initial surface concentration of probes (σP).
Conclusions
The data reported above confirm that there is in general a critical probe density threshold to achieve the most effective biomolecules recognition by immobilized probes. In particular, in the case of large HBV virus recognition by means of ssDNA strands as probes, the threshold turns to be about 4–5 × 1012 probes per cm2. Clearly, the density threshold for different probe-target couples depends on the matching of several affinity factors, including the ones controlling the diffusion of the target molecules to the probe hybridization sites, i.e., primarily the target concentration in solution and the size and conformation of the probes vs. the target to be recognized, as well as the probe–probe spacing. Accordingly, Scheme 1 reports the P1 inter-probe distance respectively for 1 × 1013 molecules per cm2 and 4.0 × 1012 molecules per cm2 for the anchored thiolated ssDNA strands. The scheme stresses the difficulty of the target of penetrating the probe layer if the probe densities are too high and the increased facility of clone penetration, turning in a selective recognition, for low-density surfaces.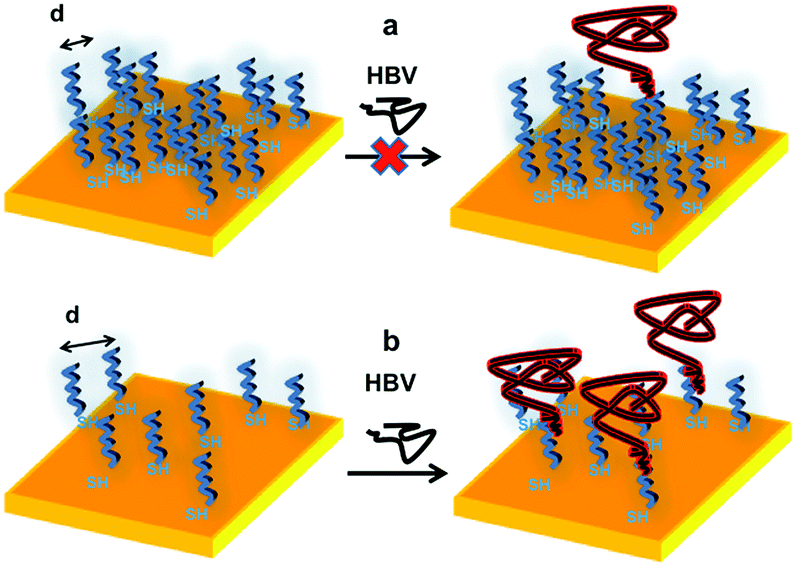 | ||
| Scheme 1 Crowding effect in the organization of thiolated ssDNA strands (21 nts) on gold surfaces: (a) high probe density surfaces, with hindered HBV hybridization; (b) low probe density surfaces, allowing efficient and selective HBV recognition. | ||
Additionally, the study revealed an important connection between the effect of ssDNA surface crowding and the kinetic step of HBV hybridization.
Indeed, to our knowledge only few literature reports are dedicated to of the study of the hybridization kinetics of ssDNA towards similar oligonucleotides,18,46–50 so that the present paper is the first model determining how the ssDNA probe density affects the association and desorption rate with a large genome, in this case the one of HBV virus. In particular, the present study paves the way to a further work aimed to develop quantitative general models for the conception and optimization of DNA chips on new conception, taking profit of a direct and selective recognition process by means of a quartz crystal microbalance sensor, working based on a single step and label free method to selectively detect genome.
The core feature of such a class of devices is clearly based on the controlled hybridization of the target biomolecules with simple linear ssDNA probes suitably immobilized on Au surfaces of the QCM resonator.
To this purpose, the control of surface density, which determines the probe accessibility, is an essential parameter for the final properties of the device and can be quantitatively controlled. Our results show that it is possible to finely control the density and, in turn, the hybridization efficiency also for very large genomes as the one of HBV. This results in a sensitivity of fmol cm−2, without the need of any amplification steps or labelling methods. These findings open intriguing prospects towards the fabrication of biosensors able to detect HBV virus in a single-step process.
In summary, the QCM-D biosensors have been shown to exhibit competitive sensitivity and fast response in detection of HBV virus combined with simple detection protocol and probe immobilization process. The described results, hence, prompt the development of an easy to use single-step and portable PoC sensors device for direct and fast HBV detection.
Acknowledgements
This work has been funded by MIUR by means of the national Program PON R&C 2007–2013, project “Hyppocrates – Sviluppo di Micro e Nano-Tecnologie e Sistemi Avanzati per la Salute dell'uomo” (PON02 00355) and PRIN 2010–2011 “Metodologie chimiche innovative per biomateriali intelligenti”.Notes and references
- J. Wang, Nucleic Acids Res., 2000, 28(16), 3011–3016 CrossRef CAS PubMed.
- M. Minunni, S. Tombelli, R. Scielzi, I. Mannelli, M. Mascini and C. Gaudiano, Anal. Chim. Acta, 2003, 481, 55–64 CrossRef CAS.
- C. Trépo, H. L. Y. Chan and A. Lok, Lancet, 2014, 384, 2053–2063 CrossRef.
- R. P. Beasley, Cancer, 1988, 61, 1942–1956 CrossRef CAS.
- J. H. Stares and E. G. Stares, Viruses and Human Disease, Academic Press, San Diego, California, 2002, p. 383 Search PubMed.
- A. S. F. Lok and B. J. McMahon, Hepatology, 2001, 34, 1225–1241 CrossRef CAS PubMed.
- H. Huang, L. Jin, X. Yang, Q. Song, B. Zou, S. Jiang, L. Sun and G. Zhou, Biosens. Bioelectron., 2013, 42, 261–266 CrossRef CAS PubMed.
- D. Paraskevis, C. Haida, N. Tassopoulos, M. Raptopoulou, D. Tsantoulas, H. Papachristou, V. Sypsa and A. Hatzakis, J. Virol. Methods, 2002, 103, 201–212 CrossRef CAS.
- W. Haasnoot, H. Gerçek, G. Cazemier and M. W. Nielen, Anal. Chim. Acta, 2007, 586, 312–318 CrossRef CAS PubMed.
- D. P. Kalogianni, T. Koraki, T. K. Christopoulos and P. C. Ioannou, Biosens. Bioelectron., 2006, 21, 1069–1076 CrossRef CAS PubMed.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- S. K. Sia and L. J. Kricka, Lab Chip, 2008, 8, 1982–1983 RSC.
- S. Conoci, A. Mascali and F. Pappalardo, RSC Adv., 2014, 4, 2845–2850 RSC.
- C. March, J. J. Manclús, Y. Jiménez, A. Arnau and A. Montoya, Talanta, 2009, 78, 827–833 CrossRef CAS PubMed.
- C. Yao, T. Zhu, J. Tang, R. Wu, Q. Chen, M. Chen, B. Zhang, J. Huang and W. Fu, Biosens. Bioelectron., 2008, 23, 879–885 CrossRef CAS PubMed.
- T. Xu, J. Miao, Z. Wang, L. Yu and C. Li, Sens. Actuators, B, 2011, 151, 370–376 CrossRef CAS PubMed.
- N. Tuccitto, N. Giamblanco, S. Ghosh, V. Spmpinato, P. Labbè, P. Dumy, S. Quici, G. Marletta, E. Defrancq and A. Licciardello, Langmuir, 2011, 27(14), 8595–8599 CrossRef CAS PubMed.
- G. Yang, Y. Gao, K. W. Lauren and R. M. Georgiadis, Nucleic Acids Res., 2006, 34(11), 3370–3377 CrossRef PubMed.
- T. Strachan and A. P. Read, in Human Molecular Genetics, Wiley-Liss, New York, 2nd edn, 1999, ch. 5, Nucleic acid hybridization assays Search PubMed.
- X. C. Zhou, L. Q. Huang and F. Y. Li Sam, Biosens. Bioelectron., 2001, 16, 85–96 CrossRef CAS.
- S. Yamaguchi, T. Shimomura, T. Tatsuma and N. Oyama, Anal. Chem., 1993, 65(14), 1925–1927 CrossRef CAS.
- H. Su and M. Thompson, Biosens. Bioelectron., 1995, 10(3/4), 329–340 CrossRef CAS.
- F. Caruso, H. Rodda, D. F. Furlong, K. Niikura and Y. Okahata, Anal. Chem., 1997, 69, 2043–2049 CrossRef CAS PubMed.
- C. Yao, T. Zhu, J. Tang, R. Wu, Q. Chen, M. Chen, B. Zhang, J. Huang and W. Fu, Biosens. Bioelectron., 2008, 23, 879–885 CrossRef CAS PubMed.
- X. Zhou, L. Liu, M. Hu, L. Wang and J. Hu, J. Pharm. Biomed. Anal., 2002, 27, 341–345 CrossRef CAS.
- H. Su, C. Sandra and M. Thompson, Biosens. Bioelectron., 1997, 12(3), 161–173 CrossRef CAS.
- A. W. Peterson, R. J. Heaton and R. M. Georgiadis, Nucleic Acids Res., 2001, 24, 5163–5168 CrossRef PubMed.
- Y. Danfeng, K. Junyoung, Y. Fang, E. P. Nielsen, E.-K. Sinner and W. Knoll, Biophys. J., 2005, 88, 2745–2751 CrossRef PubMed.
- D. Irving, P. Gong and R. Levicky, J. Phys. Chem. B, 2010, 114(22), 7631–7640 CrossRef CAS PubMed.
- J. Zeng, A. Almadidy, J. Watterson and U. J. Krull, Sens. Actuators, B, 2003, 90, 68–75 CrossRef CAS.
- Y. Ke, S. Lindsay, Y. Chang, Y. Liu and H. Yan, Science, 2008, 319, 180–183 CrossRef CAS PubMed.
- C. Lin, Y. Liu and H. Yan, Biochemistry, 2009, 48(8), 1663–1674 CrossRef CAS PubMed.
- S. Hernández, M. Venegas and R. A. Villanueva, Genome Announcements, 2014, 2(5), e01075 CrossRef PubMed.
- J. Mertens, C. Rogero, M. Calleja, D. Ramos, J. A. Martín-Gago, C. Briones and J. Tamayo, Nat. Nanotechnol., 2008, 3, 301–307 CrossRef CAS PubMed.
- G. Z. Sauerbrey, Z. Phys., 1959, 155, 206 CrossRef CAS.
- M. Yang and H. C. M. Chan, Langmuir, 1998, 14, 6121–6129 CrossRef CAS.
- Y. Xue, X. Li, H. Li and W. Zhang, Nat. Commun., 2015, 5, 4348–4357 Search PubMed.
- A. Singh, S. Snyder, L. Lee, A. P. R. Johnston, F. Caruso and Y. G. Yingling, Langmuir, 2010, 26, 17339–17347 CrossRef CAS PubMed.
- Z. F. Gao, J. B. Gao, L. Y. Zhou, Y. Zhang, J. C. Si, H. Q. Luo and N. B. Li, RSC Adv., 2013, 3, 12334–12340 RSC.
- S. B. Smith, Y. J. Cui and C. Bustamante, Science, 1996, 271, 795–799 CrossRef CAS.
- K. Rechendorff, G. Witz, J. Adamcik and G. Dietler, J. Chem. Phys., 2009, 131, 095103 CrossRef PubMed.
- G. Doni, M. D. N. Ngavouka, A. Barducci, P. Parisse, A. De Vita, G. Scoles, L. Casalisce and G. M. Pavan, Nanoscale, 2013, 5, 9988–9993 RSC.
- B. Zhao and W. J. Brittain, Prog. Polym. Sci., 2000, 25(5), 677–710 CrossRef CAS.
- G. Wu, H. Ji, K. Hansen, T. Thundat, R. Datar, R. Cote, M. F. Hagan, A. K. Chakraborty and A. Mayumdar, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1560–1564 CrossRef CAS.
- D. R. Purvis, D. Pollard-Knight and P. A. Lowe, in Commercial Biosensors, ed. G. Ramsay, Wiley-Interscience, New York, 1998, pp. 165–224 Search PubMed.
- B. P. Nelson, T. E. Grimsrud, M. R. Liles, R. M. Goodman and R. M. Corn, Anal. Chem., 2001, 73, 1–7 CrossRef CAS.
- A. W. Peterson, L. K. Wolf and R. M. Georgiadis, J. Am. Chem. Soc., 2002, 124, 14601–14607 CrossRef CAS PubMed.
- A. Vainrub and M. B. Pettitt, Biopolymers, 2003, 68, 265–270 CrossRef CAS PubMed.
- P. W. Stevens, J. Sun and D. M. Kelso, Anal. Biochem., 1999, 276, 204–214 CrossRef PubMed.
- M. F. Hagan and A. K. Chakraborty, J. Chem. Phys., 2004, 120, 4958–4968 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03467a |
| This journal is © The Royal Society of Chemistry 2015 |