
Benzylic ethers as arylcarboxy surrogates in substrate directed ortho C–H functionalisation catalysed by copper†
Abstract
A copper catalysed ortho-benzoxylation of 2-arylpyridines has been accomplished using benzylic ethers as the alternative arylcarboxy sources (ArCOO–) via sp2 C–H bond activation. The use of the Pd/TBHP catalytic system is reported to install an o-aroyl (ArCO–) moiety at the 2-arylpyridine while the Cu/TBHP combination fixes a benzoxy (ArCOO–) group at the ortho site.
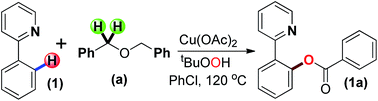
 Please wait while we load your content...
Please wait while we load your content...

