
Facile synthesis of porous Li-rich layered Li[Li0.2Mn0.534Ni0.133Co0.133]O2 as high-performance cathode materials for Li-ion batteries†
Chenwei Cao a,
Liujiang Xiab,
Kwan Lan Leunga,
Man Wanga,
Ying Liua,
Ruguang Maa,
Shiliu Yanga,
Zhouguang Lu*c and
C. Y. Chung*a
a,
Liujiang Xiab,
Kwan Lan Leunga,
Man Wanga,
Ying Liua,
Ruguang Maa,
Shiliu Yanga,
Zhouguang Lu*c and
C. Y. Chung*a
aDepartment of Physics and Materials Science, City University of Hong Kong, Hong Kong SAR, PR China. E-mail: appchung@cityu.edu.hk; Fax: +86 88018944; Tel: +86 88018966
bSchool of Metallurgical Engineering, Hunan University of Technology, PR China
cDepartment of Materials Science & Engineering, South University of Science and Technology of China, Shenzhen, PR China. E-mail: luzg@sustc.edu.cn
First published on 24th March 2015
Abstract
Lithium-rich layered metal oxides have drawn much recent attention due to their high rechargeable capacity of 250–300 mA h g−1. Herein, we report the synthesis of porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 metal oxide powders using a facile polymer-thermolysis method. X-ray powder diffractometry (XRD) results show that a well-crystallized layered structure was obtained when the calcination temperatures reach 800 °C. Pores in the range of 100–200 nm are observed using scanning electron microscopy (SEM). The porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 synthesized at 850 °C shows much superior electrochemical performance to the sample synthesized by the traditional coprecipitation-calcination method, with a high initial coulombic efficiency of 87% and initial discharge capacity of 245.4 mA h g−1 at 15 mA g−1 in the voltage window 2–4.6 V. A capacity retention of 81% was obtained after 300 cycles at 300 mA g−1. The higher capacity and improved rate performance of porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 can be predominantly attributed to enhanced Li+ intercalation kinetics resulting from the highly porous structure.
Introduction
Driven by the success of Li-ion batteries (LIBs) in the portable electronics market and the global resolution to reduce fossil fuel usage, considerable efforts are being paid to extend the use of LIBs in electrical vehicles (EVs), hybrid electrical vehicles (HEVs) and smart grids.1,2 However, current commercial cathode materials such as layered LiCoO2, LiNi0.8Co0.15Al0.05O2, LiNi0.33Co0.33Mn0.33O2, spinel LiMn2O4, and olivine LiFePO4 only have capacities around 120–200 mA h g−1, which are insufficient to meet the requirements for EV and HEV applications.3–5 Therefore, it is of great importance to develop new high-energy and high-power cathode materials.6–9Commonly denoted as xLi2MnO3·(1 − x)LiMO2 (M = Mn, Ni, Co, etc.), the lithium-rich layered metal oxides (LLOs) have attracted much attention in recent years, thanks to its high rechargeable capacity of 250–300 mA h g−1, low cost, and superior safety characteristics.10–12 The LLOs can be considered as a composite of layered LiMO2 and layered Li2MnO3 (or Li(Li1/3Mn2/3)O2), which have very similar crystal structure and can form a homogeneous solid solution. The high capacity of LLOs originates from the activation of the Li2MnO3 component when charged above 4.4–4.5 V, which transforms into electrochemically active MnO2 and improves the structural stability during cycling at the same time.13–16
Nonetheless, rate performance of the lithium-rich layer oxides is still unsatisfying due to the material's sluggish kinetics and low conductivity.17–20 Various techniques, including coprecipitation-calcination method,21–23 sol–gel process,24–26 molten salt method27 and combustion synthesis28,29 have been used to synthesize LLOs. In this study, we adopted a one-step polymer-thermolysis method to prepare LLOs. Variations of this method have been reported by Das et al.30 He et al.31 and Jiang et al.32 for the synthesis of nanocrystalline ceramics and LLOs.
Compared with other fabrication methods, the polymer-thermolysis method can produce products with good chemical homogeneity, less agglomeration, and most importantly, porous structure formed during the thermolysis process. For example, using a similar method, Jiang et al.32 has fabricated mesoporous 0.4Li2MnO3·0.6LiNi2/3Mn1/3O2 with a pore diameter of ca. 3 nm with excellent electrochemical performance. The porous structure is desirable for electrochemical applications as it creates a larger contact area between the electrode material and the electrolyte, leading to enhanced lithium diffusion and hence improving the electrochemical performance.
Experimental section
Synthesis of porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 using polymer-thermolysis (PT) method
The typical polymer-thermolysis (PT) procedure is illustrated in Scheme 1. To obtain the polymer-based precursor, firstly an aqueous solution containing stoichiometric amounts of manganese, nickel, cobalt and lithium (with 5 at% excess) nitrates was thoroughly mixed with aqueous solutions of polyvinyl alcohol (PVA) and sucrose under magnetic stirring. The molar ratio of the metal ions, PVA monomer units and sucrose was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05:2. The pH was adjusted to 1 by adding nitric acid. Then the above solution was gently heated on a hotplate until a honey-like slurry was formed. Afterwards, the slurry was slowly heated up to 250 °C in air, transforming into a fluffy voluminous carbonaceous mass, taken as the precursor. Finally, the fluffy polymer-based precursor was crushed and calcined separately at 750 °C, 800 °C, 850 °C and 900 °C for 10 hours in air. After natural cooling, the porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxide powders were obtained. For the ease of later discussion, the samples calcined at 750 °C, 800 °C, 850 °C and 900 °C were denoted as PT-750, PT-800, PT-850 and PT-900, respectively.
:0.05:2. The pH was adjusted to 1 by adding nitric acid. Then the above solution was gently heated on a hotplate until a honey-like slurry was formed. Afterwards, the slurry was slowly heated up to 250 °C in air, transforming into a fluffy voluminous carbonaceous mass, taken as the precursor. Finally, the fluffy polymer-based precursor was crushed and calcined separately at 750 °C, 800 °C, 850 °C and 900 °C for 10 hours in air. After natural cooling, the porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxide powders were obtained. For the ease of later discussion, the samples calcined at 750 °C, 800 °C, 850 °C and 900 °C were denoted as PT-750, PT-800, PT-850 and PT-900, respectively.
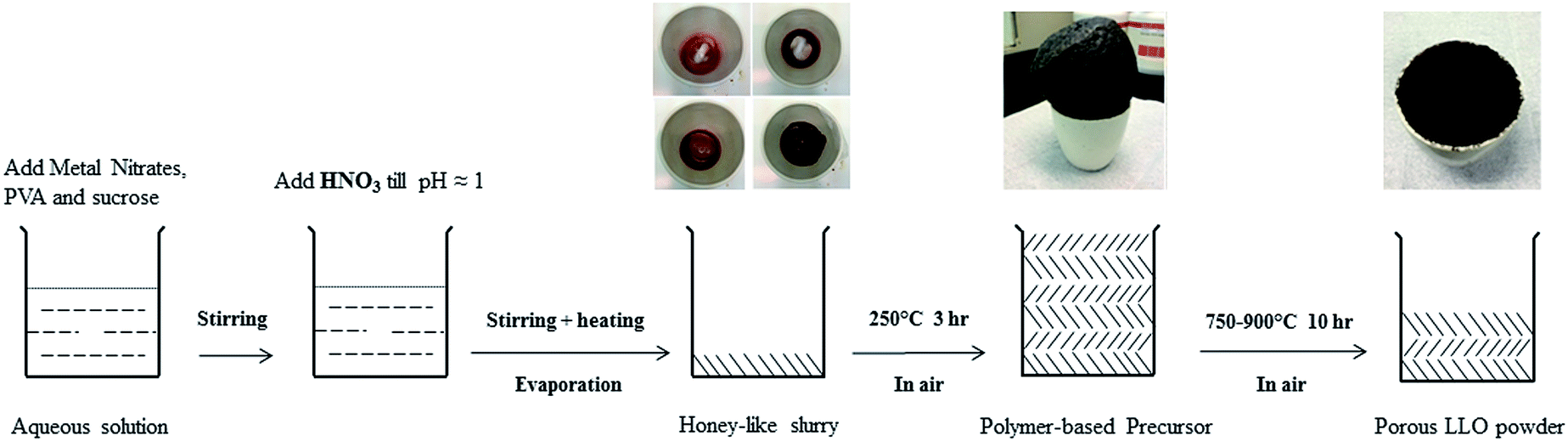 | ||
| Scheme 1 Schematic illustration of the polymer-thermolysis method for the preparation of porous LLO. | ||
Synthesis of Li[Li0.2Mn0.534Ni0.133Co0.133]O2 particles using coprecipitation-calcination (CP) method
For comparison, the Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxides were also synthesized through a two-step coprecipitation-calcination (CP) method. Transition metal hydroxide precipitates were obtained by mixing the aqueous solutions of metal nitrates and sodium hydroxide under vigorous stirring. The concentration of the solutions was 2 M. The pH was controlled at approximate 10–11. The precipitates were collected by centrifugation and dried in a vacuum chamber at 80 °C for 12 h. In the second step, the metal hydroxides and lithium hydroxide in a molar ratio of 0.8:(1.2 × 1.05) were thoroughly mixed by grinding and then calcined at 850 °C in air for 10 h. After cooling, a grayish oxide power was obtained. This sample is labelled as CP-850.
Structural and morphological characterizations
To study the crystalline structures of the as-prepared powders, X-ray diffraction (XRD) patterns in the 2θ range of 10–80° were collected using a Philips X'Pert MRD X-ray diffractometer with a Cu Kα radiation operated at 40 kV and 30 mA. The morphologies and elemental distribution of the powders were examined with a scanning electron microscope (SEM, JEOL-820) equipped with energy-dispersive X-ray spectroscopy (EDS), a field-emission SEM (Hitachi S4800) and a transmission electron microscope (TEM, CM200 FEG operated at 200 kV). Thermogravimetric analysis (TGA) was performed under oxygen atmosphere in the temperature range of 20–850 °C with a Universal V4.5A TA Instrument. The Brunauer–Emmett–Teller (BET) specific surface areas of the powders were measured from the N2 adsorption/desorption isotherms using a NOVA 1200e Surface Area and Pore Size Analyzer (Quantachrome Instruments).Electrochemical characterization
The electrochemical properties of the as-prepared Li[Li0.2Mn0.534Ni0.133Co0.133]O2 powders as cathode materials were analyzed using CR2032 coin-type half-cells. The cathode slurry was prepared by mixing Li[Li0.2Mn0.534Ni0.133Co0.133]O2 powders, acetylene black, and polyvinylidene fluoride (PVDF) binder in an 8:1:1 weight ratio, using N-methyl pyrrolidone (NMP) as solvent. The slurry was then casted onto an aluminum foil and dried at 110 °C for 12 h in a vacuum oven. Circular cathode discs were punched out from the aluminum foil. The mass of active material on each cathode disc was approximately 2 mg. The testing cells were assembled in a controlled glove box filled with argon gas, using lithium metal as the counter electrode, a layer of Celgard 2032 (Celgard, Inc., USA) as the separator, and an electrolyte composed of 1 M lithium hexafluorophosphate (LiPF6) dissolved in ethylene carbonate (EC) and dimethyl carbonate (DMC) (1:1 in volume). The fresh-assembled cells were rested for 5 hours before testing. Galvanostatic discharge–charge tests were carried out at room temperature with an Arbin Testing Workstation (BT 2000, College Station, Texas, USA). The cells were cycled in the voltage range 2–4.6 V (vs. Li+/Li) at different current densities from 0.05 to 10 C (1 C rate corresponds to a current density of 300 mA g−1). Cyclic voltammetry (CV) was performed on a Zahner IM6 electrochemical workstation. CV curves were recorded at RT between 2 and 4.6 V (vs. Li+/Li) at a scan rate of 0.1 mV s−1.
Results and discussion
The XRD patterns of the precursor and as-prepared Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxide calcined at temperatures ranging from 500 °C to 900 °C are shown in Fig. 1. | ||
| Fig. 1 XRD patterns of the precursor and Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxides synthesized at temperatures from 500 °C to 900 °C. | ||
All the strong reflections can be indexed based on a hexagonal α-NaFeO2 structure (space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m). The peaks become sharper and stronger with increasing calcination temperature. The weak minor reflections between 20–25° observed in the PT-850 and PT-900 samples can only be indexed based on the monoclinic C2/m configuration, and are characteristic of the LiMn6 cation ordering in the transition metal layers of Li2MnO3-like structures.33–36 As observed, the degree of splitting between the (006)/(102) and (108)/(110) peaks increases with increasing calcination temperature. When the calcination temperature is above 850 °C, a distinct and sharp peak-splitting is observed, indicating the formation of a well-defined layered structure, and the undesirable cation disordering between the lithium and metal ions is low.37 The intensity ratio (R) of I003/I104, an indicator of the degree of cation mixing,38 also increases from 0.672 for PT-750 to 0.879 for PT-900 (calculated after background removal). These results mean that a higher calcination temperature promotes the formation of a well-defined layered structure. No obvious difference in peak position or peak intensity is observed between sample PT-850 and CP-850.
m). The peaks become sharper and stronger with increasing calcination temperature. The weak minor reflections between 20–25° observed in the PT-850 and PT-900 samples can only be indexed based on the monoclinic C2/m configuration, and are characteristic of the LiMn6 cation ordering in the transition metal layers of Li2MnO3-like structures.33–36 As observed, the degree of splitting between the (006)/(102) and (108)/(110) peaks increases with increasing calcination temperature. When the calcination temperature is above 850 °C, a distinct and sharp peak-splitting is observed, indicating the formation of a well-defined layered structure, and the undesirable cation disordering between the lithium and metal ions is low.37 The intensity ratio (R) of I003/I104, an indicator of the degree of cation mixing,38 also increases from 0.672 for PT-750 to 0.879 for PT-900 (calculated after background removal). These results mean that a higher calcination temperature promotes the formation of a well-defined layered structure. No obvious difference in peak position or peak intensity is observed between sample PT-850 and CP-850.
SEM was performed to observe the morphology of the precursor and the as-prepared Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxide samples using PT and CP methods. The obtained SEM images are shown in Fig. 2. In the PT method, the polymer-based precursor has an irregular shape with smooth surfaces and sharp edges, with a particle size in the range of tens of micrometers. The sizes of the secondary particle of the final oxide powders are around 10 μm, and pores (∼100–200 nm across) formed between primary particles are observed on the surface. TGA results of the precursor (see Fig. S1 in the ESI†) show that upon heating above 400 °C, the polymer-precursor lost more than 80 wt% of its mass. This mass loss is most likely caused by the decomposition of the organic compounds and metal nitrates in the precursor, during which a large amount of gases, such as CO, CO2, and NO2 are released, along with the breakage of precursor particles accompanying the formation of the highly porous structure. Such porous structure may lead to an improved electrochemical performance as the increased contact area between the oxide powders and the electrolyte facilitates a faster Li-ion insertion–extraction process. The EDS mapping results (see Fig. S2†) confirms that in both the precursor and the final oxide powder, the Ni, Co and Mn ions are homogeneously distributed. As for the CP method, the morphologies of the metal hydroxide precursor and the final product are similar, with sphere-like secondary particles in the range of 1–10 μm.
 | ||
| Fig. 2 Typical SEM images of (a) the polymer-based precursor of PT method, (b) PT-750, (c) PT-800, (d) PT-850, insert: FESEM image showing the pores, (e) PT-900, (g) the metal hydroxide precursor of CP method and (h) CP-850. (f) and (i) show the TEM image of PT-850 and CP-850, respectively. | ||
TEM images of sample PT-850 and CP-850 are also shown in Fig. 2. The particle size of PT-850 (∼50–100 nm) is obviously smaller than that of CP-850 (∼100–300 nm). The Brunauer−Emmett−Teller (BET) specific surface area of sample PT-850 measured through nitrogen adsorption/desorption isotherms (see Fig. S3†) is 10.63 m2 g−1. By contrast, the specific surface area of sample CP-850 is only 2.325 m2 g−1. The small particle size and large surface area obtained by PT method are beneficial for faster Li-ion diffusion, and predict better electrochemical performance.
Fig. 3 shows the typical initial charge–discharge curves of the Li[Li0.2Mn0.534Ni0.133Co0.133]O2 samples synthesized at different temperatures at a current density of 15 mA g−1 in the voltage range of 2–4.6 V. As seen from the graph, the charge–discharge profiles are similar for all of the samples. The initial charge curve consists of a slopping a plateau region extending from 4 to 3 V, which is characteristic of the xLi2MnO3·(1 − x)LiMO2 type compounds. The sloped region corresponds to the Li+ extraction from the LiMO2 (M = Mn, Ni and Co) component, accompanied by the oxidation of Ni3+ and Co3+ ions to Ni4+ and Co3.6+, respectively.39 While the plateau region above ∼4.4 V corresponds to the Li+ extraction from the Li2MnO3 component. In the second and subsequent cycles, the plateau region in the charge curve disappears, indicating that this Li2MnO3 activation process is irreversible (see Fig. S4†).
 | ||
| Fig. 3 Initial charge–discharge curves of the Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxides calcined at different temperatures. The current density was set at 15 mA g−1. | ||
It has been shown that, although the Mn ions in Li2MnO3 cannot be further oxidized (at room temperature), the removal of Li+ is enabled by the oxidation of O2− ions, due to an overlap of the Co3+/4+:3d band with the top of the O2−:2p band, leading to the generation of oxygen vacancies and the formation of electrochemically active MnO2 phase and oxygen vacancies that serve as sites for Li-ion insertion–extraction during the following discharge–charge processes, giving rise to the anomalous high rechargeable capacity of xLi2MnO3·(1 − x)LiMO2 compounds. No obvious additional tail part is observed at the end of the first charge curves, suggesting that the amount of electrolyte decomposition in this 2–4.6 V voltage window is negligible.
The initial charge and discharge capacities, irreversible capacity loss, and coulombic efficiency of the samples are summarized in Table 1. Assuming complete Li-ion extraction from both LiMO2 and Li2MnO3 components, the theoretical capacity of Li[Li0.2Mn0.534Ni0.133Co0.133]O2 would be 377 mA h g−1. However, the actual reversible charge capacities obtained for sample PT-750, PT-800, PT-850, PT-900 and CP-850 are only 268.4, 283.9, 282, 276.5, and 285.3 mA h g−1, respectively, all significantly lower than the theoretical value. This implies that only part of the Li2MnO3 component is activated during charge in the first cycle at a cut-off voltage of 4.6 V, which is also confirmed by the CV results, which will be discussed later. Overall, samples synthesized using PT method at and above 800 °C exhibits similar charge and discharge capacities of around 280 mA h g−1 and 245 mA h g−1, respectively, as well as a high coulombic efficiency of around 87%. As for sample PT-750, a much lower charge–discharge capacity and a higher irreversible capacity loss are observed, possibly due to its poorly-formed layered structure suggested by XRD and hence a more sluggish reaction. Comparing sample PT-850 with CP-850, although they have almost identical XRD profile and initial charge–discharge voltage profiles, the discharge capacity and coulombic efficiency of CP-850 are significantly lower than that of PT-850.
| PT-750 | PT-800 | PT-850 | PT-900 | CP-850 | |
|---|---|---|---|---|---|
| Initial charge capacity (mA h g−1) | 268.4 | 283.9 | 282 | 276.5 | 285.3 |
| Initial discharge capacity (mA h g−1) | 220.3 | 242.9 | 245.4 | 245.0 | 220.5 |
| Irreversible capacity loss (mA h g−1) | 48.1 | 41.0 | 36.6 | 31.5 | 64.8 |
| Coulombic efficiency (%) | 82.1 | 85.6 | 87 | 88.6 | 77.3 |
To further understand the redox reaction processes of the obtained Li[Li0.2Mn0.534Ni0.133Co0.133]O2 powders, cyclic voltammetry (CV) tests were performed on samples PT-850 and CP-850 at a scan rate of 0.1 mV s−1 between 2 and 4.6 V for the first 5 cycles. As shown in Fig. 4, the two samples have similar CV curves. Two oxidation peaks are observed in the first charge process, one at ∼4.1 V and another at ∼4.5 V. The peak at ∼4.1 V is ascribed to the oxidation of Ni2+ to Ni4+ and Co3+ to Co3.6+, corresponding to the extraction of Li+ from the LiMO2 structure. The other peak at ∼4.5 V is related to the Li-ion extraction from the Li2MnO3 lattice accompanying with oxygen loss and possibly further oxidation of Co3.6+ to Co4+. As observed, this peak at ∼4.5 V is still present in the following cycles. On the other hand, this peak almost disappears in the second cycle when the cut-off voltage is set as 4.8 V (see Fig. S5†). Thus it can be inferred that the Li2MnO3 activation process is not easily completed in limited cycles at a cut-off voltage of 4.6 V.40 The peak at ∼4.1 V becomes broader and shifts to lower voltages, indicating an enhanced chemistry related to the Li+ extraction from the LiMO2 structure, and further confirms the transformation of Li2MnO3 into MnO2 at above 4.4 V in the first charge process. In the negative scan, there are also two reduction peaks at ∼4.5 V and ∼3.8 V in the first cycle, corresponding to the reduction of Ni4+, and Co4+ respectively. Two new broad redox peaks appear at around 2.9–3.3 V in the second cycle, and they are most likely associated with the redox reaction of Mn3+/Mn4+,13,40 indicating the formation of MnO2 after the initial Li2MnO3 activation process. The CV results agree well with the charge–discharge profiles.
 | ||
| Fig. 4 Cyclic voltammetry (CV) curves of sample PT-850 and CP-850 at a scan rate of 0.1 mV s−1 in the first five cycles. | ||
Fig. 5 shows the rate performance of the layered oxides at 15 mA g−1 to 3000 mA g−1 (0.05 C to 10 C) between 2 V and 4.6 V. The PT-850 sample again displays the best performance, with a discharge capacity of 249.7, 216.8, 208.8, 183.9, 161.5, 138.3, 122.7, 103.9, and 72.4 mA h g−1 at the current density of 15, 30, 60, 150, 300, 600, 900, 1500 and 3000 mA g−1, respectively, exceeding the capacities of sample CP-850 by about 30 mA h g−1 at all rates. This rate performance enhancement can be ascribed to the highly porous structures facilitating faster ion transport between the active particles and electrolyte.
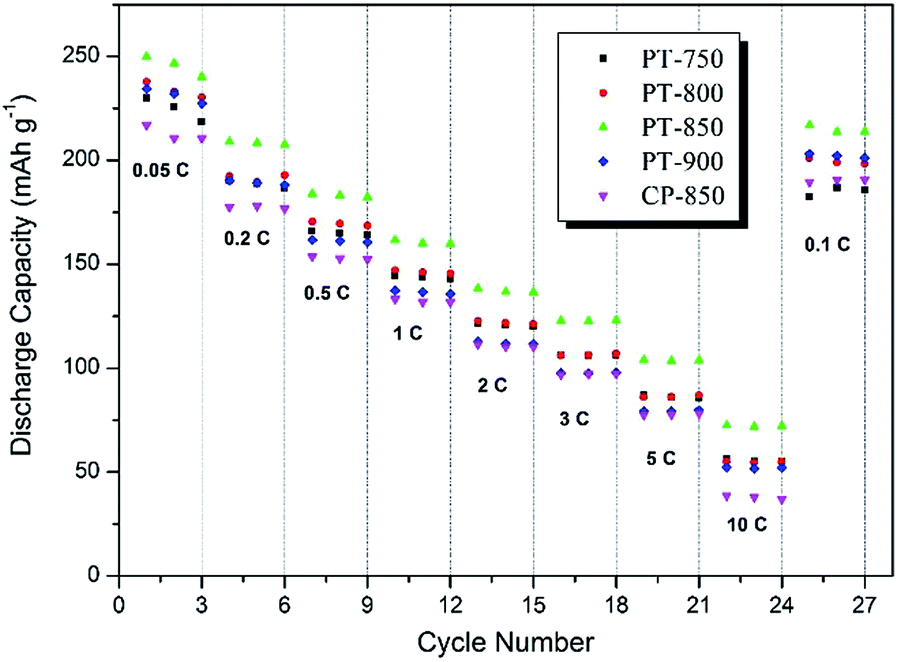 | ||
| Fig. 5 Rate performance of sample PT-850 and CP-850 from 0.05 C to 1 C between 2 and 4.6 V. | ||
Fig. 6 shows the cyclic performance of the as-synthesized samples at 300 mA g−1 (1 C) and 15 mA g−1 (0.05 C) between 2 V and 4.6 V. Although sample PT-850 exhibits the highest capacity at 1 C in the rate performance test, sample PT-800, PT-850 and PT-900 display similar cyclic performance, with a discharge capacity of 147, 150, and 157 mA h g−1 in the third cycle and a capacity retention value of approximately 81.1%, 82% and 79% after 300 cycles (with respect to the discharge capacity in the third cycle, see Fig. S6†). While for sample CP-850, only a discharge capacity of 129.2 mA h g−1 was obtained in the third cycle. Nonetheless, the capacity retention for CP-850 is approaching 90%. In the end of the 300th cycle. This effect is more significant at a low cycling current density, when cycled at 15 mA g−1 for 30 cycles, the capacity retention for PT-850 and CP-850 are 79.7% and 91.3%, respectively. The poor capacity retention of PT-850 is possibly caused by the enhanced side reaction between electrode and electrolyte, leading to Mn dissolution into the electrolyte and Mn valence state drop, which causes capacity loss.41,42 Further investigation is needed to confirm this hypothesis.
 | ||
| Fig. 6 Cyclic performance of sample PT-850 and CP-850 at (a) 300 mA g−1 (1 C) and (b) 15 mA g−1 (0.05 C) between 2 and 4.6 V. | ||
Conclusions
In summary, we have successfully synthesized high capacity porous Li[Li0.2Mn0.534Ni0.133Co0.133]O2 layered oxides using a novel polymer-thermolysis method. Their application as cathode materials for Li-ion batteries are evaluated. The best electrochemical performance was obtained in the sample synthesized at 850 °C, with a high initial discharge capacity of 245.4 mA h g−1, a high initial coulombic efficiency of 87%, a reasonably good capacity retention value of approximately 81% after 300 cycles at 300 mA g−1 and superior rate performance. Compared with the co-precipitation method, the polymer-thermolysis method produced samples with higher capacity and better rate performance thanks to the sample's very special nanoporous morphology formed by assembling of ultrafine nanoparticles. The results show that the polymer-thermolysis method could be a promising method for the synthesis of Li-rich metal oxide composite materials used as cathode materials for high energy Li-ion batteries.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21001117) and the South University of Science and Technology of China through the Talent Development Starting Fund from the Shenzhen Government. The authors would also like to thank Dr Hua Cheng and Ms Jie Zhang for their help in BET testing and analysis.Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652–657 CrossRef CAS PubMed.
- A. Manthiram, J. Phys. Chem. Lett., 2011, 2, 176–184 CrossRef CAS.
- R. Marom, S. F. Amalraj, N. Leifer, D. Jacob and D. Aurbach, J. Mater. Chem., 2011, 21, 9938–9954 RSC.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854–7863 CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- M. Hu, X. Pang and Z. Zhou, J. Power Sources, 2013, 237, 229–242 CrossRef CAS PubMed.
- K. Zaghib, A. Mauger, H. Groult, J. Goodenough and C. Julien, Materials, 2013, 6, 1028–1049 CrossRef CAS PubMed.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey, R. Benedek and S. A. Hackney, J. Mater. Chem., 2007, 17, 3112–3125 RSC.
- H. Yu and H. Zhou, J. Phys. Chem. Lett., 2013, 4, 1268–1280 CrossRef CAS.
- J. Yan, X. Liu and B. Li, RSC Adv., 2014, 4, 63268–63284 RSC.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815–A822 CrossRef CAS PubMed.
- A. R. Armstrong, M. Holzapfel, P. Novák, C. S. Johnson, S.-H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694–8698 CrossRef CAS PubMed.
- N. Yabuuchi, K. Yoshii, S.-T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404–4419 CrossRef CAS PubMed.
- Y. W. Denis and K. Yanagida, J. Electrochem. Soc., 2011, 158, A1015–A1022 CrossRef PubMed.
- A. van Bommel and J. R. Dahn, Electrochem. Solid-State Lett., 2010, 13, A62–A64 CrossRef CAS PubMed.
- H. Yu, Y. Wang, D. Asakura, E. Hosono, T. Zhang and H. Zhou, RSC Adv., 2012, 2, 8797–8807 RSC.
- M. G. Kim, M. Jo, Y.-S. Hong and J. Cho, Chem. Commun., 2009, 218–220 RSC.
- G.-Z. Wei, X. Lu, F.-S. Ke, L. Huang, J.-T. Li, Z.-X. Wang, Z.-Y. Zhou and S.-G. Sun, Adv. Mater., 2010, 22, 4364–4367 CrossRef CAS PubMed.
- Z. Lu, L. Y. Beaulieu, R. A. Donaberger, C. L. Thomas and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A778–A791 CrossRef CAS PubMed.
- Y. Wang, N. Sharma, D. Su, D. Bishop, H. Ahn and G. Wang, Solid State Ionics, 2013, 233, 12–19 CrossRef CAS PubMed.
- X. Yang, X. Wang, G. Zou, L. Hu, H. Shu, S. Yang, L. Liu, H. Hu, H. Yuan, B. Hu, Q. Wei and L. Yi, J. Power Sources, 2013, 232, 338–347 CrossRef CAS PubMed.
- J. Li, L. Wang, L. Wang, J. Luo, J. Gao, J. Li, J. Wang, X. He, G. Tian and S. Fan, J. Power Sources, 2013, 244, 652–657 CrossRef CAS PubMed.
- J. Shojan, V. R. Chitturi, L. Torres, G. Singh and R. S. Katiyar, Mater. Lett., 2013, 104, 57–60 CrossRef CAS PubMed.
- O. Toprakci, H. A. K. Toprakci, Y. Li, L. Ji, L. Xue, H. Lee, S. Zhang and X. Zhang, J. Power Sources, 2013, 241, 522–528 CrossRef CAS PubMed.
- C. Yu, G. Li, X. Guan, J. Zheng and L. Li, J. Alloys Compd., 2012, 528, 121–125 CrossRef CAS PubMed.
- S. J. Shi, J. P. Tu, Y. Y. Tang, Y. X. Yu, Y. Q. Zhang, X. L. Wang and C. D. Gu, J. Power Sources, 2013, 228, 14–23 CrossRef CAS PubMed.
- C. Ghanty, S. Chatterjee, R. N. Basu and S. B. Majumder, J. Electrochem. Soc., 2013, 160, A1406–A1414 CrossRef CAS PubMed.
- R. N. Das, Mater. Lett., 2001, 47, 344–350 CrossRef CAS.
- W. He, J. Qian, Y. Cao, X. Ai and H. Yang, RSC Adv., 2012, 2, 3423–3429 RSC.
- Y. Jiang, Z. Yang, W. Luo, X.-L. Hu, W.-X. Zhang and Y.-H. Huang, J. Mater. Chem., 2012, 22, 14964–14969 RSC.
- M. M. Thackeray, S.-H. Kang, C. S. Johnson, J. T. Vaughey and S. A. Hackney, Electrochem. Commun., 2006, 8, 1531–1538 CrossRef CAS PubMed.
- J. Bréger, M. Jiang, N. Dupré, Y. S. Meng, Y. Shao-Horn, G. Ceder and C. P. Grey, J. Solid State Chem., 2005, 178, 2575–2585 CrossRef PubMed.
- K. A. Jarvis, Z. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614–3621 CrossRef CAS.
- H. Koga, L. Croguennec, P. Mannessiez, M. Ménétrier, F. Weill, L. Bourgeois, M. Duttine, E. Suard and C. Delmas, J. Phys. Chem. C, 2012, 116, 13497–13506 CAS.
- T. A. Arunkumar, E. Alvarez and A. Manthiram, J. Electrochem. Soc., 2007, 154, A770–A775 CrossRef CAS PubMed.
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2002, 48, 145–151 CrossRef CAS.
- T. A. Arunkumar, Y. Wu and A. Manthiram, Chem. Mater., 2007, 19, 3067–3073 CrossRef CAS.
- H. Yu, H. Kim, Y. Wang, P. He, D. Asakura, Y. Nakamura and H. Zhou, Phys. Chem. Chem. Phys., 2012, 14, 6584–6595 RSC.
- J. Zheng, M. Gu, J. Xiao, P. Zuo, C. Wang and J.-G. Zhang, Nano Lett., 2013, 13, 3824–3830 CrossRef CAS PubMed.
- X. Lin, J. Park, L. Liu, Y. Lee, A. M. Sastry and W. Lu, J. Electrochem. Soc., 2013, 160, A1701–A1710 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03445k |
| This journal is © The Royal Society of Chemistry 2015 |