DOI:
10.1039/C5RA03430B
(Paper)
RSC Adv., 2015,
5, 36798-36808
Screening of modified CaO-based catalysts with a series of dopants for the supercritical water gasification of empty palm fruit bunches to produce hydrogen
Received
25th February 2015
, Accepted 13th April 2015
First published on 13th April 2015
Abstract
Catalytic supercritical water gasification (SCWG) of empty palm fruit bunches (EFB) was carried out using bulk and modified CaO-based catalysts with several selected dopants. The catalysts were prepared via a wet impregnation method and characterized using X-ray diffraction (XRD), N2 adsorption (BET), temperature programmed reduction (TPR-H2) and temperature programmed desorption (TPD-CO2). The catalytic reactions were performed using 0.3 g of EFB with 5 wt% of the catalysts in 8 mL of deionized water at 380 °C. The results show that addition of the catalysts into the EFB SCWG reaction improves the overall gas yield and hydrogen selectivity. Furthermore, the catalysts after reduction were found to be more active than the unreduced catalysts in which the presence of metallic Ni enhances the gasification reaction. The addition of bulk CaO into the reaction improves the hydrogen yield (50.6 mmol mL−1) when compared to the reactions conducted in the absence of a catalyst (41.3 mmol mL−1) while the addition of 5 wt% of Ni-doped CaO shows a further improvement (57 mmol mL−1). However, the addition of secondary dopants into Ni–CaO shows significant elevation in the hydrogen concentration. Among the catalysts studied, Zn-doped Ni–CaO gave the highest hydrogen yield (105.7 mmol mL−1) due to its increased promotional effects on the water gas shift reaction. The effect of the dopants on the CaO catalyst in the EFB SCWG reaction is discussed in detail.
1. Introduction
Growing interest in hydrogen as an alternative energy source has widened its utilization in many applications. The use of hydrogen is considered a progressive approach to combat greenhouse gas emissions, especially the release of CO2 from the combustion of fossil fuels. Hydrogen is a renewable and green fuel source when compared to current gaseous fuels, where its combustion only releases heat and pure water as the final product.1 However, elemental hydrogen does not exist naturally and the current supply is produced by the conversion of fossil fuels; a process that is considered unsustainable due to the excessive emission of greenhouse gases and pollutants to the environment.2 The substantial interest in hydrogen as a potential fuel in the near future has led to efforts to produce it from various sources such as the electrolysis of water, water pyrolysis, thermal pyrolysis and reforming of organic compounds, and via methanation of microorganisms.3 The generation of hydrogen from biomass is a very promising method to produce green energy due to the zero CO2 emissions from the process as it is believed to be consumed by biomass growth via photosynthesis.4 However, the major obstacle in biomass conversion is the high moisture content of the feedstock, which is unfit for many conventional thermochemical conversion techniques (such as gasification, pyrolysis and combustion) and require a pre-drying step before the actual production process.
Supercritical water gasification is a well-established technique that is suitable for wet biomass conversion into hydrogen rich product gas. The technique exhibits high solid conversion and apparently reduces tar and char formation during the reaction.5 The specific features of water under supercritical conditions (temperature < 273 °C and pressure ∼ 22 MPa) are its low dielectric constant, viscosity and a reduced number of hydrogen bonds that are weakened by the harsh environment and significantly alter the basic properties of water.6 Under these circumstances, water behaves like a homogenous non-polar solvent with a high solubility capacity that favors the dissolution of organic compounds and reduces the mass transfer limitations of the reaction.7,8 As well as being the reaction medium, water acts as a catalyst in the SCWG process whereby the ionic products (H+ and OH−) obtained from the dissociation of water molecules can facilitate acid or base catalyzed reactions including the hydrolysis and pyrolysis of organic compounds.3,8 However, not all the compounds in biomass are completely transformed into gases during the SCWG reaction. There are various factors involved in achieving complete gasification such as feedstock concentration, reaction temperature and retention time. The formation of tar and char is unavoidable during the SCWG reaction due to the polymerization of the dissolved compounds. Likewise, the catalyst free-SCWG reaction leads to higher yields of CO due to the low tendency of the water gas shift reaction and high reaction temperatures are essential to attain acceptable conversion.9
Generally, elevated reaction temperatures and pressures are the driving force for H2 production in the SCWG reaction. However, the use of a catalyst improves the reaction in many aspects such as avoiding the necessity of extreme reaction conditions, enhancing the selectivity of the reaction towards H2 and eliminating the formation of tar and char. Furthermore, the presence of a catalyst is linked with its ability to perform C–C bond cleavage, enhancement of the water gas shift reaction and minimize C–O bond breaking that favors the methanation reaction.10 In general, supercritical water gasification catalysts are classified into homogenous catalysts (KOH, NaOH, Na2CO3 and K2CO3) and heterogeneous catalysts (activated carbon and supported transition metals). Homogenous catalysts mainly promote the water gas shift reaction when compared to heterogeneous catalysts, which are considered more suitable for the SCWG reaction due to their higher selectivity and recyclability.3 A detailed review on the heterogeneous catalysts used in the SCWG reaction has been reported by Azadi et al. and are classified into activated carbon, supported and unsupported transition metal (Ni, Ru, Pt and Pd) and metal oxide (CaO, ZrO2 and CeO2) catalysts.9
In this paper the effect of tri-metal oxide catalysts produced from bulk CaO that have been added with primary (Ni) and secondary dopants (La, Mg, Ba, Nd, Na, K, Zn, Co and Fe) was investigated in the SCWG reaction of empty palm fruits bunches. CaO was found to be active in enhancing the yield of hydrogen and promote carbon gasification in the SCWG reaction.11,12 Therefore, modified CaO catalysts and the effect of dopants on their catalytic activity during the SCWG reaction were studied in detail.
2. Experimental
2.1 Sample preparation
EFB were collected from a local palm oil extraction mill in Johor. The fresh bunches were chopped into smaller pieces and dried for 2 weeks. The dried sample was shredded and sieved using a 250 μm siever. Ultimate analysis and the theoretical moles of the EFB are given in Table 1.13,14
Table 1 Ultimate analysis and theoretical mol EFB
| Feedstock |
C |
H |
N |
S |
O |
| Elemental composition (%) |
50.2 |
7.1 |
0.8 |
0.01 |
41.9 |
| Theoretical mol (mmol g−1) |
12.6 |
11 |
— |
— |
7.9 |
2.2 Catalyst preparation and characterization
The catalysts were synthesized using a wet impregnation method. Bulk CaO (Sigma Aldrich, 24856-8) was used as the base catalyst and was added with the primary dopant (Ni(NO3)2·6H2O) and a series of secondary dopants [La(NO3)2, Mg(NO3)2·6H2O, Ba(NO3)2, Nd(NO3)3·6H2O, NaNO3, KNO3, Zn(NO3)2, Co(NO3)2·6H2O and Fe(NO3)3] obtained from Sigma-Aldrich. 5 wt% of Ni and 5 wt% of the selected secondary dopant were diluted together in deionized water. The salt solutions were added to the bulk CaO (57 nm), stirred for 6 h at room temperature and dried overnight at 110 °C. The obtained precursor catalysts were crushed into powders and calcined at 900 °C for 6 h under an air environment. Furthermore, the catalysts were reduced in 5% H2 balanced in a flow of argon (20 mL min−1) at 700 °C for 3 h. The prepared catalysts are listed in Table 2. The physico-chemical properties of the catalysts were characterized by X-ray diffraction (XRD), Brunner–Emmet–Teller (BET) surface area, temperature programmed reduction (TPR) and temperature programmed desorption (TPD-CO2). The X-ray diffraction patterns of the catalysts were analyzed at ambient temperature using a Shimadzu diffractometer XRD6000 equipped with a Philips glass diffraction X-ray tube broad focus 2.7 kW type to produce Cu-Kα radiation. The BET surface area of the catalysts was obtained using a Thermo Finnigan Sorptomatic 1900 series model instrument via N2 adsorption/desorption isotherms patterns at −196 °C. TPR-H2 and TPD-CO2 analyses were performed using a Thermo Finnigan TPDRO 1100 apparatus equipped with a thermal conductivity detector with 20 mg of sample. For TPR analysis the sample was pre-treated under an N2 environment and analysis continued in a flow of 5% H2/argon (25 mL min−1) in the temperature range 50–900 °C. TPD-CO2 was carried out with pre-treatment of the catalysts under an N2 environment at 30–400 °C for 20 min and CO2 gas purged for absorption over the catalyst for 1 h at 50 °C. Then, the CO2 absorbed catalysts were heated from 50–900 °C and the desorbed gas analyzed using the TCD signals.
Table 2 The physicochemical properties of the prepared catalysts
| Sample |
BET surface area (m2 g−1) |
Pore volume (BJH analysis) (cm3 g−1) |
CaO crystallite size (nm) |
NiO crystallite size (nm) |
| CaO |
5.5 |
0.0211 |
57.0 |
— |
| 5NiO–CaO |
7.2 |
0.0223 |
51.3 |
50.4 |
| 5La2O3/5NiO–CaO |
4.7 |
0.0144 |
53.0 |
55.3 |
| 5MgO/5NiO–CaO |
4.9 |
0.0139 |
57.0 |
46.7 |
| 5BaO/5NiO–CaO |
3.1 |
0.0096 |
59.0 |
53.6 |
| 5Nd2O5/5NiO–CaO |
2.6 |
0.0079 |
53.0 |
57.2 |
| 5Na2O/5NiO–CaO |
1.9 |
0.0049 |
48.0 |
39.9 |
| 5K2O/5NiO–CaO |
2.5 |
0.0068 |
62.0 |
42.8 |
| 5ZnO/5NiO–CaO |
2.9 |
0.0098 |
53.9 |
43.6 |
| 5CoO/5NiO–CaO |
3.3 |
0.0101 |
51.0 |
42.8 |
| 5Fe2O3/5NiO–CaO |
7.0 |
0.0208 |
57.0 |
21.9 |
2.3 Catalytic studies
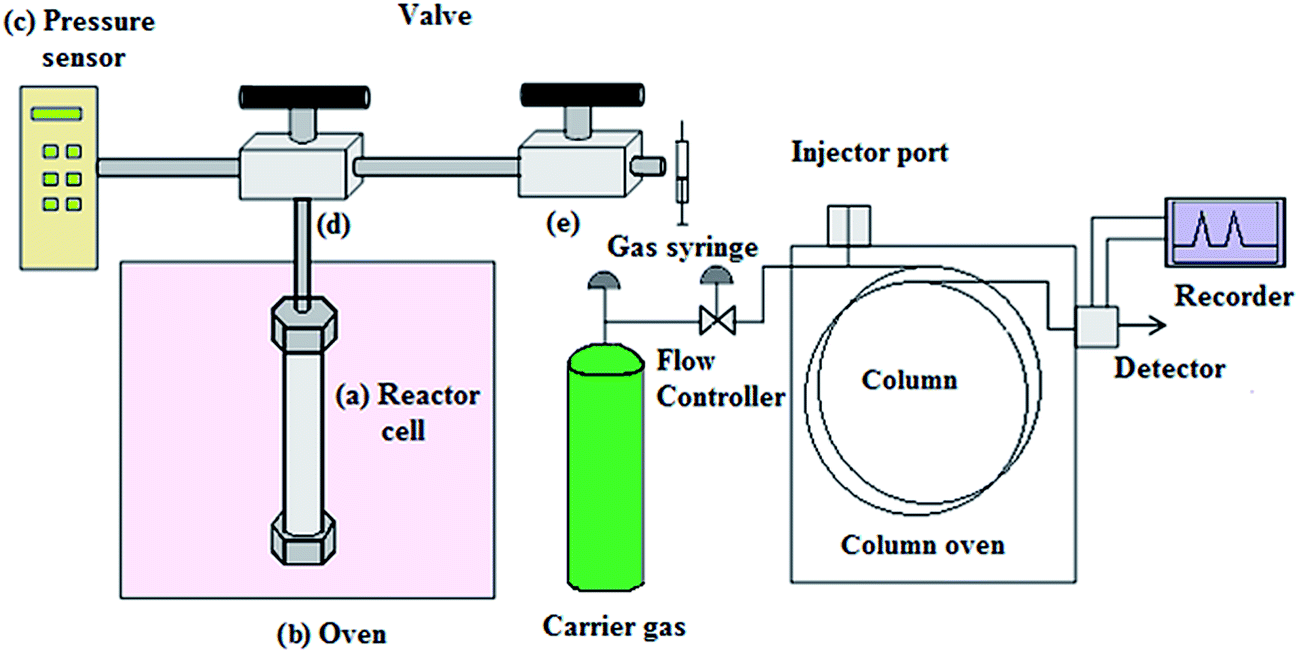
The SCWG reaction was performed in a custom made reactor using stainless steel tubing and 13 mL reactor cell. About 0.3 g of the EFB sample was added into the reactor cell with an initial 5 wt% of catalyst and 8 mL of deionized water. The reactor cell was sealed tightly and fixed to the reactor frame as shown in Fig. 1. The reactor frame was placed inside a gas chromatography (GC) oven used as a heating chamber that can be heated over a temperature range of 30–400 °C. The reactor cell was heated to 380 °C at a heating rate of 10 °C min−1 and held at this temperature for 8 min. Then, the reactor cell was cooled down to 30 °C and the valves opened intermittently to collect the small fraction of the products gases (0.25 mL) using a gas tight syringe straight from the reactor cell. The collected product gas was injected into a GC (Agilent) equipped with two columns (Porapak Q and Mole sieve) and a thermal conductivity detector to measure the concentration of the product gases.
 |
| | Fig. 1 A schematic diagram of the supercritical water gasification reactor. | |
3. Results and discussion
3.1 Catalyst characterization
3.1.1 XRD patterns of the unreduced and reduced catalysts. The prepared catalysts were characterized using several techniques including XRD, BET, TPD-CO2 and TPR-H2. The catalysts were also reduced in a 5% H2/argon flow (20 mL min−1) at 700 °C for 3 h to compare their catalytic activity with the unreduced catalysts. Fig. 2(a and b) shows the X-ray diffraction patterns of the reduced and unreduced catalysts. In both catalysts, three major peaks were found at 32.2°, 37.4° and 54.3° corresponding to the presence of CaO. In addition, several small intensity peaks were also observed at 18°, 47° and 50.8° associated to Ca(OH)2 due to hydration of CaO with atmospheric moisture. Apart from this, there are several peaks with very low intensity related to the dopants added to CaO. However, due to the very low amount of loading and the limitations of instrument detection, most of the dopants present were not observed in the XRD patterns. Nevertheless, there are a few peaks observed for the unreduced catalyst at 33.5°, 43.3° and 43.1° corresponding to Ca2Fe2O5,15 CoNiO and NiO phase formation, respectively. After reduction several low intensity peaks also appeared at 43.01°, 43.06° and 43.11° corresponding to Mg3NiO4, Ni8Zn2O and CaNiO as a result of strong interactions between the dopants added on the CaO surface. Based on the XRD results, the average cluster size of the CaO and NiO was calculated using the Debye–Scherrer equation (Table 2). The average crystal size of CaO was in the range of 48–62 nm and NiO shows a wide range of crystal size between 21–57 nm, which were influenced by the dispersion and possible agglomeration that occurred during the calcination step.
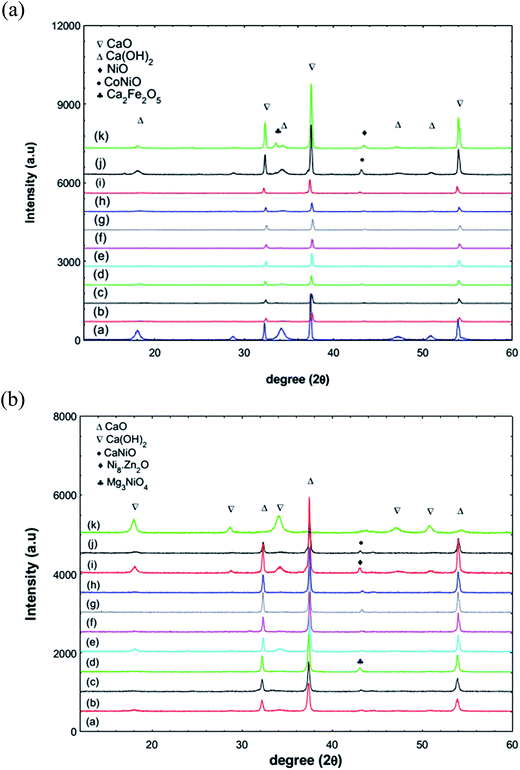 |
| | Fig. 2 (a): XRD patterns of the unreduced catalysts. (a) CaO, (b) 5Ni–CaO, (c) 5La/5Ni–CaO, (d) 5Mg/5Ni–CaO, (e) 5Ba/5Ni–CaO, (f) 5Nd/5Ni–CaO, (g) 5Na/5Ni–CaO, (h) 5K/5Ni–CaO, (i) 5Zn/5Ni–CaO, (j) 5Co/5Ni–CaO, (k) 5Fe/5Ni–CaO. (b): XRD patterns of the reduced catalysts. (a) CaO, (b) 5Ni–CaO, (c) 5La/5Ni–CaO, (d) 5Mg/5Ni–CaO, (e) 5Ba/5Ni–CaO, (f) 5Nd/5Ni–CaO, (g) 5Na/5Ni–CaO, (h) 5K/5Ni–CaO, (i) 5Zn/5Ni–CaO, (j) 5Co/5Ni–CaO, (k) 5Fe/5Ni–CaO. | |
3.1.2 BET surface area. The BET surface area and average pore diameter of the prepared catalysts are presented in Table 2. Bulk CaO was found to be less porous with a measured surface area of ∼5.5 m2 g−1 and average pore diameter of >50 nm in the macroporous range. Therefore, upon the addition of the dopants they were predicted to be dispersed on the surface of CaO and could influence the surface area of the catalysts. Based on the results shown in Table 2, the surface area and pore volumes were slightly changed upon the addition of the dopants caused by several possible factors such as partial coating and blockages of the small pores on the CaO surface by the additives and changes in the mass density or interaction of CaO with the dopants.16 In agreement with the literature, the CaO with added dopants showed a reduction in surface area with the exception of the NiO–CaO and Fe2O3/NiO–CaO catalysts that show a slight increase in surface area. The increment was possibly caused by the deformation on the CaO crystal structures created by the incorporated dopants, which cause cracks and the formation of new pores on the surface of the support.17 Furthermore, the addition of Fe tends to reduce NiO sintering where a smaller crystal size (21.9 nm) was observed when compared to the other catalysts and indirectly increased the overall surface area.18
3.1.3 Temperature programmed desorption-CO2 analysis. The basicity of the catalysts shows a high influence on the overall catalytic activity and product yield. Generally, basic supports have a higher tendency towards hydrogen production when compared to alkane formation, which is favored by acidic supports during biomass gasification.19 Fig. 3 displays the TPD-CO2 profiles and the total amount of CO2 desorption relative to temperature is shown in Table 3. The basic properties of CaO were altered upon addition of the different dopants. The CO2 desorption nature was improved and the catalysts show stronger basic strength (above 600 °C) that slightly varies upon the addition of the dopants. It was found that the highest CO2 desorption was observed with the MgO added NiO–CaO catalysts. The effect of MgO as a dopant has been reported in the literature and was shown to promote the basic strength of the support material, which enhances the CO2 absorption, facilitates coke removal by providing ample surface oxygen and also tends to increase the dispersion of nickel on the support.20 A slight difference in the basic strength of each catalyst was observed at the temperature where the CO2 desorption peak occurred. Therefore, the strength of the basicity of the catalyst was slightly improved upon addition of the dopants when compared to bulk CaO.
 |
| | Fig. 3 Temperature programmed desorption-CO2. (a) CaO, (b) 5Ni–CaO, (c) 5La/5Ni–CaO, (d) 5Mg/5Ni–CaO, (e) 5Ba/5Ni–CaO (f) 5Nd/5Ni–CaO, (g) 5Na/5Ni–CaO, (h) 5K/5Ni–CaO, (i) 5Zn/5Ni–CaO, (j) 5Co/5Ni–CaO, (k) 5Fe/5Ni–CaO. | |
Table 3 The basicity and reducibility of the catalysts
| Sample |
CO2 desorbed (μmol g−1) |
Peak temperature (°C) |
H2 consumption (μmol g−1) |
Peak temperature (°C) |
| CaO |
1593.7 |
615 |
338.1 |
610.3 |
| 5NiO–CaO |
2079.3 |
623 |
812.9 |
555.4 |
| 5La2O3/5NiO–CaO |
1200.0 |
651 |
784.2 |
558.7 |
| 5MgO/5NiO–CaO |
2904.6 |
649 |
163.6 |
611.1 |
| 5BaO/5NiO–CaO |
1701.4 |
616 |
684.6 |
610.3, 726.8 |
| 5Nd2O5/5NiO–CaO |
999.4 |
623 |
799 |
561.2 |
| 5Na2O/5NiO–CaO |
1395.0 |
639 |
1220.1 |
547.0, 722.7 |
| 5K2O/5NiO–CaO |
2244.1 |
620 |
1531.9 |
576.2 |
| 5ZnO/5NiO–CaO |
1315.1 |
621 |
501.4 |
561.2, 764.3 |
| 5CoO/5NiO–CaO |
2899.8 |
654 |
1704.9 |
576.2 |
| 5Fe2O3/5NiO–CaO |
2820.2 |
661 |
2719.7 |
631.1, 705.2 |
3.1.4 Temperature programmed reduction-H2. Fig. 4 shows the TPR profiles of the synthesized catalysts in a partial hydrogen flow balanced by argon (25 mL min−1) at 700 °C for 3 h. Variances in the TPR profile of each catalyst are noticed as a function of the added dopants, which influence the reducibility and its interaction with CaO. Table 3 summarizes the H2 consumption and its relative temperature range based on the strength of the dopant interactions. Bulk CaO was difficult to reduce due to the strong Ca–O bond. However, a very small reduction peak was seen (338.1 μmol g−1) at elevated temperature (610.3 °C) probably caused by a small amount of Ca–O bond reduction on the surface. Furthermore, the addition of NiO into CaO shows a notable shift in the reduction peaks. It was reported in the literature that unsupported bulk NiO was reduced at temperatures in the range of 400–418 °C.21 However, the NiO–CaO reduction peak was observed at 555.4 °C, which indicates the presence of a chemical interaction between NiO and CaO. Furthermore, higher H2 consumption (812.9 μmol g−1) was observed for the NiO–CaO catalyst when compared to bulk CaO indicating the reduction of Ni2+ to Ni0. Apart from this, upon the addition of the secondary dopants almost all the major reduction peaks occurred in the range of 550–650 °C corresponding to the strong chemical interactions of the dopants with CaO. Some of the catalysts including Ba, Na, Zn and Fe doped NiO–CaO exhibit two peaks, whereby the second peak appeared at temperatures above 700 °C. This peak was possibly due to several factors such as the reduction of the secondary dopant or the stronger interaction of NiO with the added dopants, which shifts the peak to a higher temperature. Among the catalysts, the highest H2 consumption was observed for the Fe-doped catalyst followed by Co, K and Na. Moreover, smaller NiO particles have been reported to be difficult to reduce and require higher temperatures for reduction to occur.22 This was confirmed by the smaller crystal size of NiO in the Fe-doped catalyst when compared to the other catalysts studied.
 |
| | Fig. 4 Temperature programmed reduction-H2. (a) CaO, (b) 5Ni–CaO, (c) 5La/5Ni–CaO, (d) 5Mg/5Ni–CaO, (e) 5Ba/5Ni–CaO, (f) 5Nd/5Ni–CaO, (g) 5Na/5Ni–CaO, (h) 5K/5Ni–CaO, (i) 5Zn/5Ni–CaO, (j) 5Co/5Ni–CaO, (k) 5Fe/5Ni–CaO. | |
3.2 The catalytic supercritical water gasification reaction
The supercritical water gasification reaction has received a lot of attention as a feasible method for the conversion of high moisture content biomass into product gas rich in hydrogen. The effects of the catalysts on the process are vital in order to enhance the hydrogen yield and carbon conversion. Therefore, bulk and modified CaO with the addition of several dopants as listed in Table 2 were used as catalysts in the EFB supercritical water gasification reaction. The preliminary screening of the catalysts was carried out to study the role of CaO and the effect of the added dopants on the H2 yield and carbon conversion efficiency. It has been reported in the literature that biomass SCWG reactions involves a complex reaction mechanism where thermal decomposition of the feedstock occurs via pyrolysis and hydrolysis reactions.23 Generally, the composition of the SCWG product gas includes H2, CO2, CO and CH4 and was significantly influenced by the various reaction mechanisms occurring in the process such as water gas shift (eqn (1)), steam reforming (eqn (2)) and methanation reactions (eqn (3)). However, the addition of the catalyst shows a significant improvement in the desired product yield along with carbon conversion. Among the complex reaction mechanisms occurring in the process, the water gas shift reaction plays an important role in elevating the overall hydrogen yield produced in the SCWG reaction via CO conversion in the presence of steam into H2 and CO2. Generally, catalyst-free reactions tend produce higher amounts of CO as a consequence of a reduced number of water gas shift reactions (eqn (1)) and higher reaction temperatures are often needed to achieve an adequate carbon conversion.9 Therefore, the addition of a suitable catalyst that favors high H2 production and significant carbon conversion is very importance in the SCWG reaction.| | |
CHxOy + (1 − y)H2O → CO + (1 − y + x/2)H2
| (2) |
In the preliminary studies, 5 wt% of CaO was added into the mixture (0.3 g EFB and 8 mL of deionized water) and the reaction carried out at 380 °C with a range of reaction times from 8–32 min. The effects upon adding the catalyst on the composition of the product gas obtained from the EFB SCWG reaction are presented in Fig. 5. When compared to the uncatalyzed reaction, CaO loading slightly improved the hydrogen concentration. Furthermore, increasing the reaction time from 8 to 32 min shows steady improvement in the hydrogen production due to a mild increase in the water gas shift reaction with reaction time. However, increasing the reaction time also enhanced methane production, which was possibly produced from both the methanation reaction and functional group cleavage in the feedstock. Besides, a very low amount of CO implies the tendency of CaO to promote the water gas shift reaction in the SCWG reaction based on several factors such as its CO2 absorption capacity. However, apart from CO2 absorption, Zhang et al. found that the addition of CaO in the coal water slurry SCWG reaction catalyzed the gasification, water gas shift and reforming reactions.12 In addition, the basic nature of the catalysts enhanced the water gas shift reaction, propelled by the cyclic coordination of the intermediate anions. Initially, the carbonate ion formed reacts with water to produce hydroxide ions and carbon dioxide. Subsequently, the hydroxide ions combine with carbon monoxide to form formate ions. The unstable formate decomposes back into carbonate and yields formaldehyde, which tends to degrade by releasing hydrogen.24 Therefore, based on the promising catalytic activity of CaO, further improvements on the catalyst were carried out by introducing 5 wt% of nickel. Generally, metallic Ni was used as the dopant supported on various materials such as Al2O3, MgO, CeO2, activated carbon and SiO2.25–29 Ni has been widely studied as a promising metal catalyst to promote the SCWG reaction via enhancing the water gas shift reaction, C–C bond breaking and improving the overall carbon conversion.30,31 However, the nickel-based catalyst was also found to indirectly promote the methanation reaction (eqn (3)) by catalyzing C–O bond cleavage, which reduces the hydrogen yield.31 Therefore, 5 wt% of a secondary dopant was added into the Ni-doped CaO catalysts to enhance the gasification reaction and selectivity towards a higher hydrogen yield. The effects of several secondary dopants (La, Mg, Ba, Nd, Na, K, Zn Co and Fe) was investigated to identify the promising metal dopant that could be used to elevate the desired product yield. In addition, the influence of the added dopants on CaO was investigated in both the unreduced and reduced materials.
 |
| | Fig. 5 Catalytic supercritical water gasification of EFB upon the addition of CaO. | |
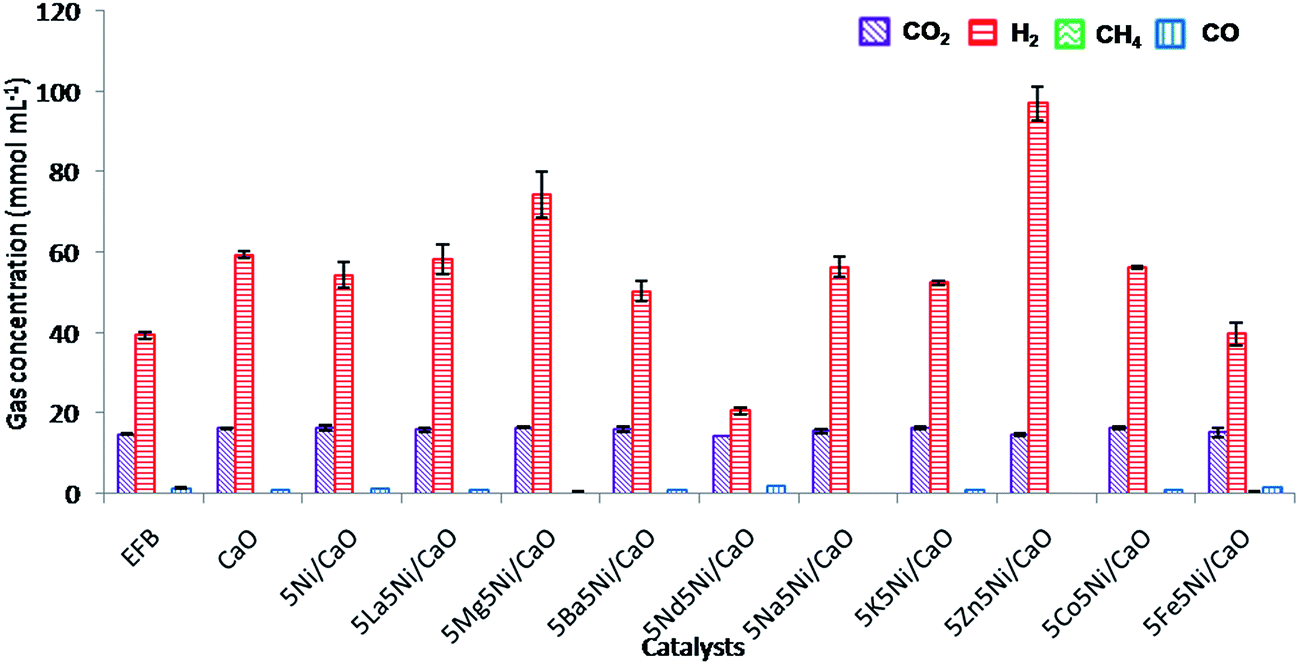
The composition of the product gas from the EFB SCWG reactions using the unreduced catalysts are shown in Fig. 6. The unreduced catalyst with 5 wt% NiO on CaO shows a slight drop in the hydrogen yield when compared to bulk CaO. It was found that the addition of NiO did not improve the catalytic activity and lowered the tendency of the water gas shift reaction as indicated by the higher amount of CO produced during the reaction. Also, Azadi et al. reported that NiO shows a lower catalytic activity than its reduced form in the SCWG reaction.9 Furthermore, the presence of dopants on the surface of CaO could also reduce the absorption of CO2 due to their interactions with the active sites of CaO involved in CO2 capture. A reduction in the amount of CO2 produced was essential in the SCWG reaction mechanism to shift the reaction equilibrium to the right and drive the water gas shift reaction towards higher H2 and CO2 production.
 |
| | Fig. 6 Product gas composition of the EFB catalytic supercritical water gasification reaction (unreduced catalysts). | |
Furthermore, the addition of secondary dopants on the NiO–CaO catalyst shows a wide effect on the overall composition of the product gas and especially on the water gas shift reaction. It was found that upon the addition of La, Ba, Nd, Na, K, Co, and Fe the production of hydrogen production was not improved. However, among the secondary dopants, MgO and ZnO were found to be promising dopants that significantly increase hydrogen production when compared to the other dopants investigated. The addition of ZnO on the NiO–CaO catalysts significantly improved the overall hydrogen yield (97.1 mmol mL−1) due to its high promotional effects on the water gas shift reaction. The lowest amount of CO produced was observed in the gas composition as a result of a great boost in the water gas shift reaction due to the presence of ZnO in the catalysts.
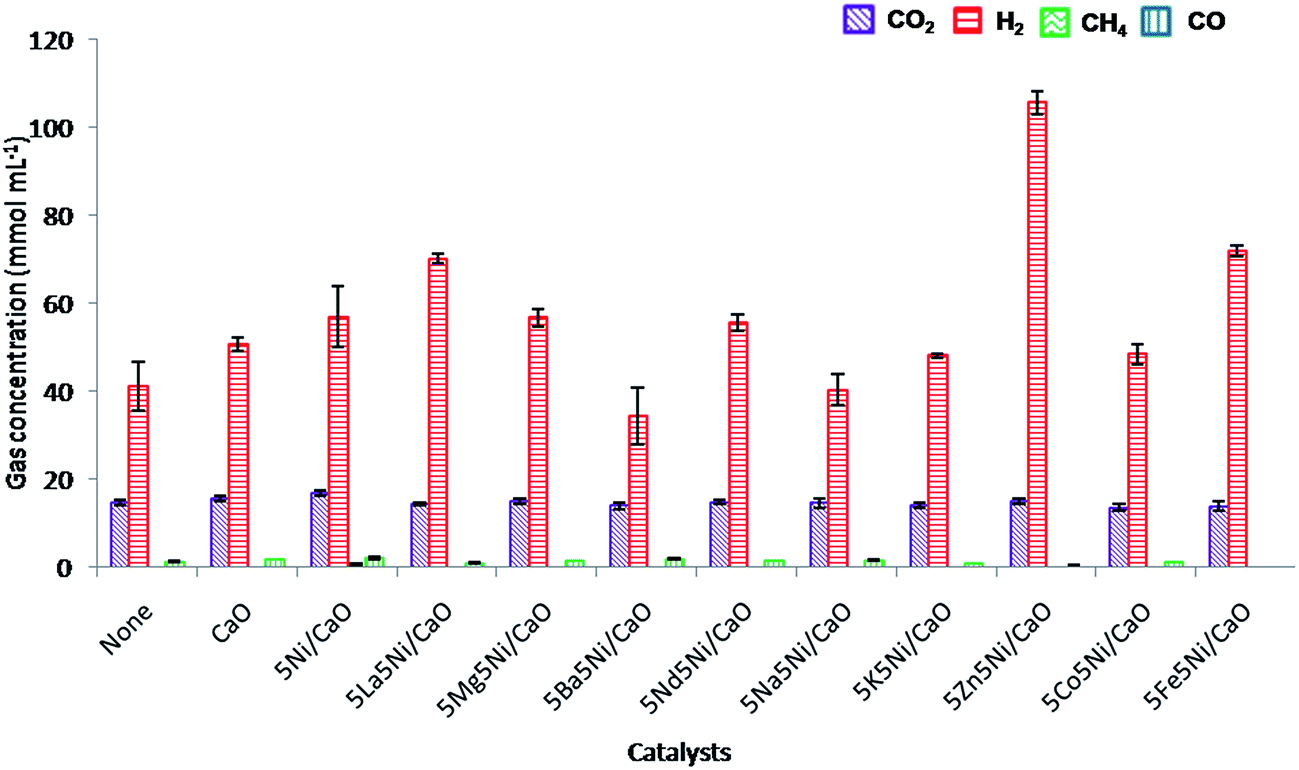
In comparison, the reduced catalysts show a notable improvement in the gas composition as shown in Fig. 7. Furthermore, the effect on the carbon conversion of EFB during the SCWG reaction in the presence of the synthesized catalysts is summarized in Table 4. After reduction, the presence of metallic Ni on CaO shows a slight improvement when compared to the NiO–CaO catalysts in terms of carbon gasification and hydrogen concentration. It was found that the carbon concentration in the gas phase increased from 18.4 mmol mL−1 to 19.4 mmol mL−1 and the hydrogen concentration improved to 57 mmol mL−1 compared to 54.4 mmol mL−1. Reduced nickel was found to be active in catalyzing several reactions including C–C bond cleavage, steam reforming and the water gas shift reaction. However, metallic nickel also favors the unwanted methanation reaction by assisting C–O bond breaking and reduces the overall production of hydrogen.32 In addition, secondary dopants including La, Zn and Fe on the Ni–CaO catalysts enhanced the catalytic performance and selectivity towards the desired product yield.
 |
| | Fig. 7 Product gas composition of the EFB catalytic supercritical water gasification reaction (reduced catalysts). | |
Table 4 A summary of the catalytic supercritical water gasification of EFBsa
| Catalysts |
Reaction time (min) |
Carbon concentration in gas phase (mmol mL−1) |
CO2/CH4 ratio |
| Unreduced catalyst |
Reduced catalyst |
Unreduced catalyst |
Reduced catalyst |
| Reaction temperature = 380 °C. Reaction time = 8 min. R refers to reduced catalyst. |
| 5NiO–CaO |
8 |
18.4 |
19.4 |
30.3 |
26.2 |
| 5La2O3/5NiO–CaO |
8 |
17.5 |
16.0 |
26.5 |
23.1 |
| 5MgO/5NiO–CaO |
8 |
17.7 |
16.9 |
26.2 |
26.7 |
| 5BaO/5NiO–CaO |
8 |
17.7 |
16.2 |
29.4 |
28.3 |
| 5Nd2O5/5NiO–CaO |
8 |
17.0 |
16.9 |
29.1 |
24.6 |
| 5Na2O/5NiO–CaO |
8 |
16.7 |
16.7 |
30.1 |
31.1 |
| 5K2O/5NiO–CaO |
8 |
17.9 |
15.4 |
31.3 |
29.4 |
| 5ZnO/5NiO–CaO |
8 |
15.5 |
15.8 |
28.2 |
26.5 |
| 5CoO/5NiO–CaO |
8 |
18.1 |
15.2 |
30.1 |
30.4 |
| 5Fe2O3/5NiO–CaO |
8 |
17.4 |
14.8 |
30.9 |
25.4 |
| Catalysts |
Reaction time (min) |
Carbon concentration (mmol mL−1) |
CO2/CH4 ratio |
| None |
8 |
16.5 |
21.8 |
| CaO |
8 |
18.0 |
30.4 |
| CaO |
16 |
15.8 |
14.6 |
| CaO |
24 |
16.8 |
13.6 |
| CaO |
32 |
15.6 |
10.1 |
| 5ZnO/5NiO–CaO (R) |
16 |
16.3 |
14.8 |
| 5ZnO/5NiO–CaO (R) |
24 |
17.1 |
12.4 |
| 5ZnO/5NiO–CaO (R) |
32 |
16.3 |
8.2 |
Among the dopants, the zinc-doped catalyst significantly enhanced the production of hydrogen when compared to the other catalysts prepared with unreduced catalysts. Both ZnO and CaO are hardly reduced under a flow of H2 at the annealing temperature (700 °C) due to the strong Zn–O and Ca–O bonds.33 Therefore, the catalyst was predicted to be in the form of metallic Ni with ZnO supported on CaO. However, the strong interactions of Ni with ZnO tended to produce a Ni8Zn2O phase, which was difficult to reduce as indicated by the TPR-H2 spectra. The presence of both metallic Ni and Ni8Zn2O on the CaO surface could promote the SCWG reaction including the water gas shift reaction, reforming reaction and C–C cracking steps. Therefore, the synergistic effect of Ni and ZnO on the CaO catalysts provides a great promotional effect on the water gas shift reaction. It has been reported in the literature that interface sites between nickel and zinc significantly promote the water gas shift reaction. Tang and Chuang proposed two possible reaction mechanisms for the water gas shift reaction favored by a combination of both nickel and zinc (eqn (4)–(13)).33 Moreover, the ability of CaO to capture the CO2 produced was an additional advantage of the catalysts.
Pathway 1
| | |
H2O + 2*(i) ↔ H* + OH*
| (5) |
| | |
OH*(i) + CO*(i) ↔ COOH*(i) + *(i)
| (6) |
| | |
COOH*(i) + *(i) ↔ CO2*(i) + H*(i)
| (7) |
Pathway 2
| | |
H2O + *(i) ↔ H2 + O*
| (10) |
| | |
CO + O*(i) ↔ CO2 + *(i)
| (11) |
| | |
CO + NiO* ↔ CO2 + Ni*
| (12) |
| | |
H2O + Ni* ↔ H2 + NiO*
| (13) |
Based on the proposed mechanism, pathway 1 was more likely to occur during the catalytic SCWG reaction due to the involvement of interface sites formed between nickel and zinc. As we reported earlier, after the catalysts were reduced the presence of metallic Ni and the Ni8Zn2O phase were observed in the XRD patterns of the catalyst. This observation elucidates the strong interaction of NiO with ZnO, which was difficult to reduce at 700 °C and was retained after the catalysts were reduced. Therefore, Ni and the Ni8Zn2O phase play an important role in catalyzing the SCWG reaction including C–C cracking, reforming and the water gas shift reactions. Besides, H2O dissociation was found to be more favorable at the oxide–metal interphase than on the metallic or oxide surfaces. Several suitable oxides have been reported including CeO2−x, TiO2−x, MoO3−x and ZnO1−x that are capable of forming a metal–oxide interface upon addition of a metal. The bi-functional properties of the catalyst include H2O adsorption and dissociation on the oxide surface. CO absorption takes place on the nearby metal to produce intermediates that are decomposed to H2 and CO2.34 Therefore, the formation of a Ni8Zn2O solid solution provides an ideal condition for the WGS reaction to progress under the SCWG reaction and increase hydrogen selectivity.
However, only a slight improvement in the carbon conversion was observed in the reaction using the reduced catalysts. There are several factors that indirectly influence the overall carbon conversion such as reaction temperature, type of feedstock and the catalyst used. Generally, 350–600 °C is regarded as a low reaction temperature and complete carbon conversion difficult to achieve even with the addition of a catalyst.35 Generally, supported noble metal catalysts such as Ru, Rh and Pt (low loading) are used in the low temperature catalytic SCWG reaction, which withstand early catalyst deactivation and achieve significant carbon conversion when compared to Ni-based catalysts.36 Among the noble metals, ruthenium exhibits a very high catalytic activity in the low temperature SCWG reaction due to several factors, similar to other noble metals, including higher metal dispersion, lower surface mobility and mild reduction temperature.3,9 However, the use of expensive catalysts has potential drawbacks and should be considered in order to keep the overall operational cost reasonable for further scale up of the process. Apart from this, EFB is real biomass waste containing hemicellulose, cellulose and lignin that are interlinked together in a complex structure. Therefore, decomposition of the EFBs requires harsher reaction conditions when compared with most of the model compounds (glucose, cellulose, lignin and xylan) used in previous SCWG studies.14 Likewise, metallic nickel also tends to be deactivated during the reaction via the formation of thermally stable tar, which can cover the active sites on the catalyst. Therefore, under the reaction environment used in this investigation, it was difficult to achieve a high carbon conversion using real biomass (EFB) with the Ni-based catalysts. However, ZnO-doped Ni–CaO shows a very high selectivity towards hydrogen due to its high catalytic activity for enhancing the water gas shift reaction.
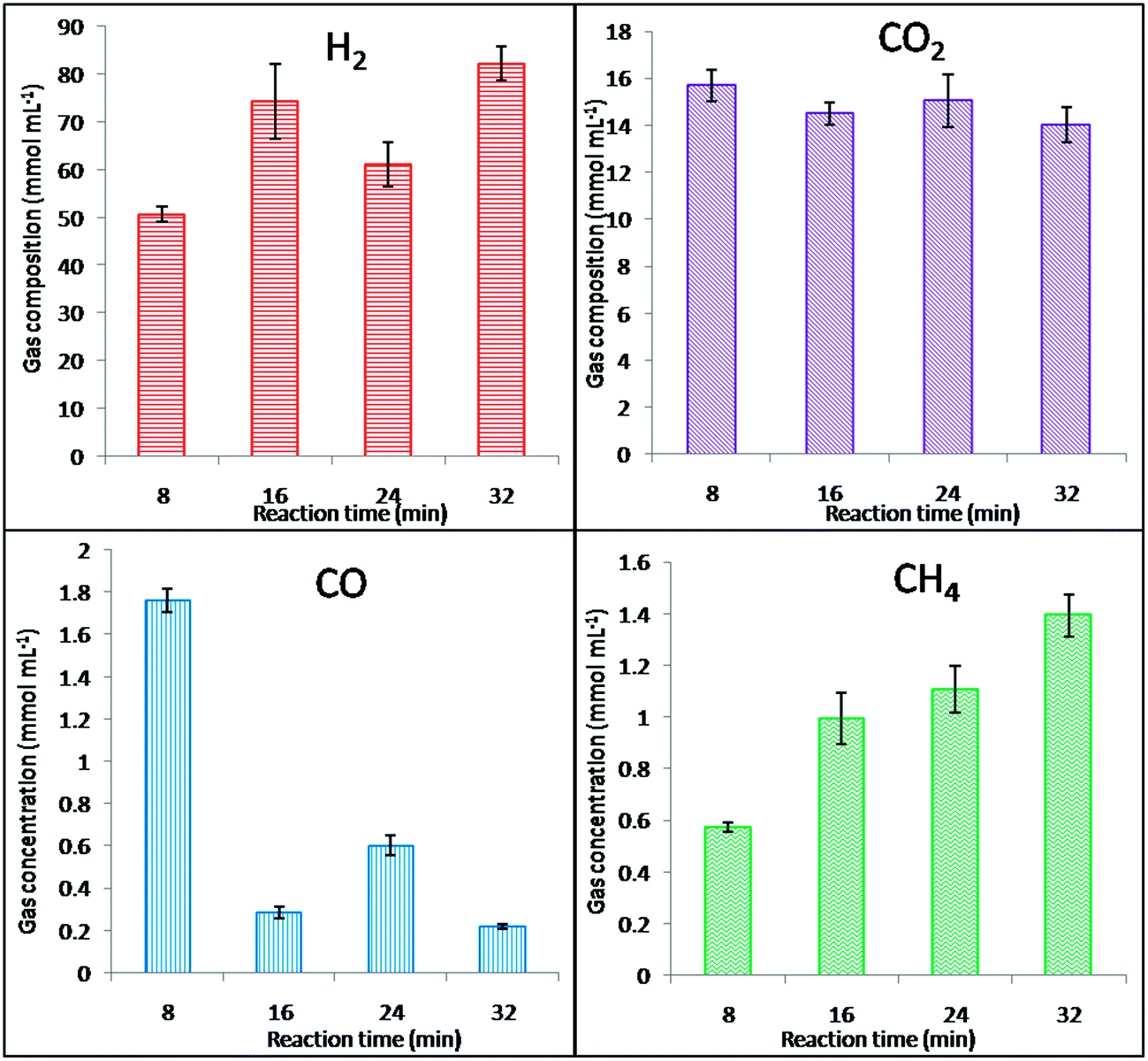
Based on our previous studies on the EFB SCWG reaction in the absence of a catalyst, increasing the reaction time did not show any notable improvement in the composition of the product gas. Decomposition of EFB under supercritical conditions tends to release the dissolved compounds, which can agglomerate and form a thermally stable tar under the reaction conditions used. This phenomenon prevents further degradation of the tarry compounds even with an increase in reaction time. However, we also investigated the effect of reaction time in the presence of Ni–ZnO/CaO. Fig. 8 shows the product gas composition in the Ni–ZnO/CaO added EFB SCWG reaction at different reaction times in the range of 8–32 min. It was found that the production of hydrogen was slightly increased while no significant difference was observed for CO2. However, CO and CH4 show drastic changes in their production whereby the amount of CO formed was reduced due to the water gas shift reaction. In addition, the amount of CH4 formed quadrupled upon increasing the reaction time from 8 to 32 min, which was caused by both the methanation reaction and functional group cleavage. In addition, metallic Ni was reported to be active in promoting both the water gas shift reaction and the unwanted methanation reaction that consumes the hydrogen produced. However, increasing the reaction time did not increase the carbon conversion and an almost constant carbon concentration was observed with time (Table 4). The presence of a residue in the reactor cell indicates the low carbon conversion, probably due deactivation of metallic Ni on the catalysts caused by tar formation, which favors C–C bond breaking. Therefore, an 8 min reaction was taken into consideration as the optimum condition for the catalytic reaction where the effect of reaction time was not significant in terms of achieving a higher hydrogen yield and carbon conversion.
 |
| | Fig. 8 The effect of the reaction time SCWG of EFB using Ni–ZnO/CaO. | |
4. Conclusions
We have shown that catalytic supercritical water gasification of EFB is a promising technique, suitable for feedstocks that contain a high moisture content. The development of an appropriate catalyst for the SCWG reaction was necessary to achieve product gas that was rich with hydrogen. Therefore, CaO was used as a catalyst and was added with 5 wt% Ni and 5 wt% of a secondary dopant (La, Mg, Ba, Nd, Na, K, Zn, Co and Fe) to enhance its catalytic activity. Among the catalysts prepared, the Zn-doped Ni–CaO catalysts were found to be very active and produced the highest hydrogen yield when compared to the other catalysts used in this study. In comparison, the reduced catalysts were more active than their corresponding unreduced catalysts due to the presence of metallic Ni that can facilitate several reactions including C–C bond breaking, methanation and water gas shift reactions. However, the presence of ZnO in the catalyst shows a tremendous improvement in the water gas shift reaction when compared to the other secondary dopants studied. The synergistic effect of the Ni8Zn2O phase and metallic Ni on CaO in the reduced catalyst enhances the SCWG reaction and its selectivity towards hydrogen production. However, a lower carbon conversion was also observed during the reaction as a result of the moderate reaction conditions (including reaction temperatures of 380 °C) and the complex structure of real biomass (EFB), which was thermally stable under the reaction conditions used. In addition, the effect of reaction time (8–32 min) with the selected Ni–ZnO/CaO catalyst in the SCWG reaction shows less improvement in the gas composition and carbon conversion. Furthermore, increasing the reaction time favors the production of CH4, which was possibly caused by methanation and functional group cleavage. Therefore, 8 min was considered as an optimum reaction time for the EFB SCWG reaction using the reduced Ni–ZnO/CaO catalysts. Among the catalysts, the ZnO-doped Ni–CaO catalyst was found to be a promising candidate for the EFB SCWG reaction and produced the highest hydrogen yield due to the enhancement of the water gas shift reaction.
References
- H. Balat and E. Kirtay, Int. J. Hydrogen Energy, 2010, 35, 7416–7426 CrossRef CAS PubMed.
- P. N. Sheth and B. V. Babu, Int. J. Hydrogen Energy, 2010, 35, 10803–10810 CrossRef CAS PubMed.
- Y. Guo, S. Z. Wang, D. H. Xu, Y. M. Gong, H. H. Ma and X. Y. Tang, Renewable Sustainable Energy Rev., 2010, 14, 334–343 CrossRef CAS PubMed.
- E. A. Youssef, M. B. I. Chowdhury, G. Nakhla and P. Charpentier, Int. J. Hydrogen Energy, 2010, 35, 5034–5042 CrossRef CAS PubMed.
- J. Yanik, S. Ebale, A. Kruse, M. Saglam and M. Yuksel, Fuel, 2007, 86, 2410–2415 CrossRef CAS PubMed.
- Y. Lu, L. Guo, X. Zhang and C. Ji, Int. J. Hydrogen Energy, 2007, 37, 3177–3185 CrossRef PubMed.
- Y. Calzavara, C. Joussot-Dubien, G. Boissonnet and S. Sarrade, Energy Convers. Manage., 2005, 46, 615–631 CrossRef CAS PubMed.
- P. E. Savage, J. Supercrit. Fluids, 2009, 47, 407–414 CrossRef CAS PubMed.
- P. Azadi and R. Farnood, Int. J. Hydrogen Energy, 2011, 36, 9529–9541 CrossRef CAS PubMed.
- S. N. Reddy, S. Nanda, A. K. Dalai and J. A. Kozinski, Int. J. Hydrogen Energy, 2014, 39, 6912–6926 CrossRef CAS PubMed.
- L. Cheng, Z. Rong and B. I. Ji-Cheng, J. Fuel Chem. Technol., 2007, 35(3), 257–261 CrossRef CAS.
- R. Zhang, W. Jiang, L. Cheng, B. Sun, D. Sun and J. Bi, Int. J. Hydrogen Energy, 2010, 35, 11810–11815 CrossRef CAS PubMed.
- S. Sivasangar, Y. H. Taufiq-Yap, Z. Zainal and K. Kitagawa, Int. J. Hydrogen Energy, 2013, 38, 16011–16019 CrossRef CAS PubMed.
- S. Sivasangar, Z. Zainal, A. Salmiaton and Y. H. Taufiq-Yap, Fuel, 2015, 143, 563–569 CrossRef CAS PubMed.
- D. Hirabayashi, T. Yoshikawa, K. Mochizuki, K. Suzuki and Y. Sakai, Catal. Lett., 2006, 110(3–4), 269–274 CrossRef CAS PubMed.
- Y. Li, X. Wang, C. Xie and C. Song, Appl. Catal., A, 2009, 357, 213–222 CrossRef CAS PubMed.
- Y. H. Taufiq-Yap, S. Sivasangar and A. Salmiaton, Energy, 2012, 47, 158–165 CrossRef CAS PubMed.
- A. S. Bambal, K. S. Vecchio and R. J. Cattolica, Ind. Eng. Chem. Res., 2014, 53, 13656–13666 CrossRef CAS.
- R. R. Davda, J. W. Shabaker, G. W. Huber, R. D. Cortright and J. A. Dumesic, Appl. Catal., B, 2005, 56, 171–186 CrossRef CAS PubMed.
- K. Y. Koo, H. S. Roh, Y. T. Seo, D. J. Seo, W. L. Yoon and S. B. Park, Appl. Catal., A, 2008, 340, 183–190 CrossRef CAS PubMed.
- R. Brown, M. E. Cooper and D. A. Whan, Appl. Catal., 1982, 3, 177–186 CrossRef CAS.
- B. Mile, D. Stirling, M. A. Zammitt, A. Lovell and M. Web, J. Catal., 1988, 114, 217–229 CrossRef CAS.
- I. Ronnlund, L. Myreen, K. Lundqvist, J. Ahlbeck and T. Westerlund, Energy, 2011, 36, 2151–2163 CrossRef PubMed.
- D. C. Elliott, R. T. Hallen and L. J. Sealock Jr, Ind. Eng. Chem. Prod. Res. Dev., 1983, 22, 431–435 CrossRef CAS.
- S. Li, Y. Lu, L. Guo and X. Zhang, Int. J. Hydrogen Energy, 2011, 36, 14391–14400 CrossRef CAS PubMed.
- T. Furusawa, T. Sato, H. Sugito, Y. Miura, Y. Ishiyama, M. Satoa, N. Itoh and N. Suzuki, Int. J. Hydrogen Energy, 2007, 32, 699–704 CrossRef CAS PubMed.
- Y. J. Lu, L. J. Guo, C. M. Ji, X. M. Zhang, X. H. Hao and Q. H. Yan, Int. J. Hydrogen Energy, 2006, 31, 822–831 CrossRef CAS PubMed.
- I. G. Lee, Int. J. Hydrogen Energy, 2011, 36, 8869–8877 CrossRef CAS PubMed.
- T. Minowa and S. Inoue, Renewable Energy, 1999, 16, 1114–1117 CrossRef CAS.
- Y. Lu, Y. Zhu, S. Li, X. Zhang and L. Guo, Biomass Bioenergy, 2014, 67, 125–136 CrossRef CAS PubMed.
- P. Azadi, E. Afif, F. Azadi and R. Farnood, Green Chem., 2012, 14, 1766–1777 RSC.
- M. Inaba, K. Murata, M. Saito and I. Takahara, Energy Fuels, 2006, 20, 432–438 CrossRef CAS.
- C. W. Tang and S. S. C. Chuang, Int. J. Hydrogen Energy, 2014, 39, 788–797 CrossRef CAS PubMed.
- J. A. Rodriguez, S. Ma, P. Liu, J. Hrbek, J. Evans and M. Perez, Science, 2007, 318, 1757–1759 CrossRef CAS PubMed.
- Y. Matsumura, T. Minowa, B. Potic, S. R. A. Kersten, W. Prins, W. P. M. van Swaaij, B. V. D. Beld, D. C. Elliott, G. G. Neuenschwander, A. Kruse and M. J. Antal Jr, Biomass Bioenergy, 2015, 29, 269–292 CrossRef PubMed.
- A. A. Peterson, F. Vogel, R. P. Lachance, M. Froling Jr, M. J. Antal and J. W. Tester, Energy Environ. Sci., 2008, 1, 32–65 CAS.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.