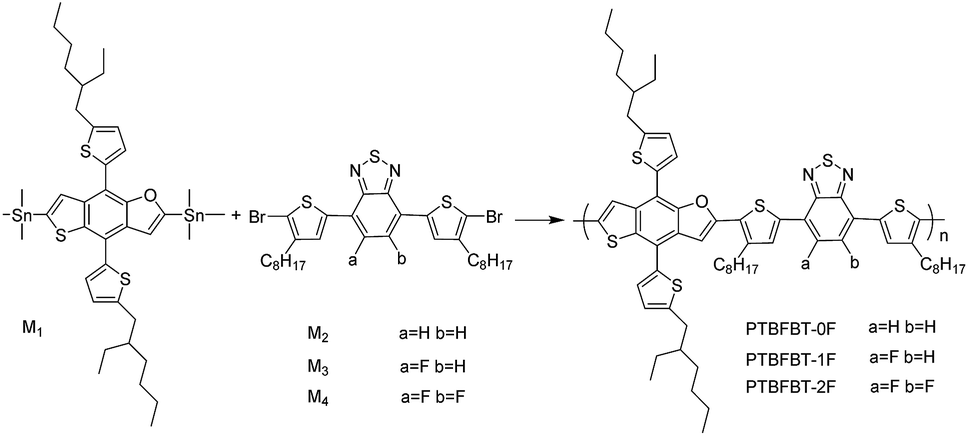
DOI:
10.1039/C5RA03405A
(Paper)
RSC Adv., 2015,
5, 30145-30152
Effect of fluorination on the performance of poly(thieno[2,3-f]benzofuran-co-benzothiadiazole) derivatives†
Received
24th February 2015
, Accepted 16th March 2015
First published on 16th March 2015
Abstract
In order to shed light on the effects of different numbers of fluorine atoms in donor–acceptor (D–A) conjugated polymers on the photophysics and photovoltaic properties, three polymers named PTBFBT-0F, PTBFBT-1F, PTBFBT-2F were synthesized and thoroughly investigated. The nonfluorinated benzothiadiazole (BT) polymer (PTBFBT-0F) has a highest occupied molecular orbital (HOMO) energy level of −4.98 eV and a low bandgap of 1.64 eV. When one of the hydrogen atoms of the BT unit was substituted by a fluorine atom (PTBFBT-1F), a small blue-shift in UV-Vis absorption and a lower HOMO energy level of −5.11 eV were observed, thus a similar Voc and Jsc were obtained. Nevertheless, the FF of PTBFBT-2F was further improved from 46% to 53% due to the relatively higher and balanced electron/hole charge transport mobility of 1.83 × 10−5 cm2 V−1 s−1 and 1.52 × 10−5 cm2 V−1 s−1. Using a Ca/Al top electrode, devices based on PTBFBT-0F, PTBFBT-1F, PTBFBT-2F as electron donor showed increasing power conversion efficiencies (PCE) of 3.0%, 3.6% and 4.2%, respectively. Furthermore, replacing Ca with a zirconium acetylacetonate film (ZrAcac) as the cathode buffer layer (CBL), a PCE of 5% with PTBFBT-2F as the donor was obtained.
Introduction
Bulk heterojunction (BHJ) polymer solar cells (PSCs) sandwich a blended layer of conjugated polymer donor and fullerene derivative acceptor between a transparent ITO positive electrode and a low work function metal negative electrode. Since the first report of a BHJ PSC 1995,1 the PCE of the PSCs has been improved gradually by designing new donor and acceptor materials as well as new device structures, and recently PCEs exceeding 10% have been reported.2–6 Although high PCE has been obtained, improved efficiency and stability are still needed to enhance these cells towards realizing commercialization. Therefore, research on device structures tends to focus on conventional devices, tandem configurations4,7 and inverted devices.8–12 Development of new photovoltaic materials has mainly focused on creating new donor–accepter (D–A) conjugated polymers with alternating electron-rich and electron-deficient units along the polymer main chain, because their bandgap and energy levels can be easily tuned by controlling the intramolecular charge transfer (ICT) from the D unit to the A unit.13 In order to obtain higher PCE, donor materials with improved charge transport properties, a narrower band gap (Eg), deeper HOMO energy levels and superior solubility as well as miscibility with fullerenes are urgently needed. Because these parameters are trade-offs, it is a great challenge to design and synthesize a material to meet all these requirements.
From previous work it can be seen that the F atom has many merits for improving photovoltaic performance, for example, (a) its strong electronegativity induces greater intramolecular and intermolecular interactions which can optimize the morphology of the active layer;14–16 (b) its strong electron-withdrawing nature can lower the HOMO energy level, and thereby increase the Voc of PSCs;17,18 (c) the induced dipole may also facilitate charge transfer with less geminate recombination by reducing coulombic interaction between electron and hole.19,20 However, the effect of the number of substituted fluorine atoms on PCE is rarely reported.
The BT unit has been very popular in the design of photovoltaic materials due to its excellent optical and electrochemical properties. The synthesis of a conjugated polymer with a fluorinated BT unit was first reported by You et al., and its improved performance was demonstrated compared to the unsubstituted BT moiety.21 Other groups have subsequently reported the use of the fluorinated BT unit which in general resulted in an improved photovoltaic performance.22–26 Therefore, the fluorinated BT unit has great potential as the electron accepting unit in the construction of high efficiency donor materials. Meanwhile, there is also an urgent need to find a suitable donor unit. Our previous materials based on the thieno[2,3-f]benzofuran (TBF) unit have shown an efficient PCE up to 6.4%.27 Herein, in order to explore the effects of different numbers of F atoms in the BT unit, we chose a D–A polymer backbone based on TBF as the donor unit and differently fluorinated BTs as acceptor unit with thiophene as a π bridge.18,28 Accordingly, three D–A polymers, named PTBFBT-0F, PTBFBT-1F and PTBFBT-2F were designed and synthesized (Scheme 1). We intend to explore the changes of Voc, fill factor (FF) and Jsc of the above-mentioned polymers due to the introduction of one or two F atoms. After one fluorine atom was introduced into the BT unit, the copolymer exhibited a low-lying HOMO energy level and a small blue-shift in the UV-Vis absorption resulting in a higher Voc (from 0.63 V to 0.77 V) and a lower Jsc (from 11.18 mA cm−2 to 10.28 mA cm−2) as compared with those of the non-fluorinated analogue. At the same time, the FF was improved from 43% to 46%. Upon the introduction of a second fluorine atom into BT, the FF was further improved from 46% to 53% due to the relatively higher and balanced electron/hole charge transport mobility, but almost constant Jsc (from 10.28 mA cm−2 to 10.54 mA cm−2) and Voc (from 0.77 V to 0.75 V) were obtained resulting from the similar HOMO level and bandgap. We also found that the electrochemical and optical properties of these polymers with PC71BM could be affected by fluorination of the polymer backbone. Therefore, introducing different numbers of F atoms into the acceptor unit in conjugated polymers is a promising method to effectively tune their properties for optoelectronic applications.
 |
| | Scheme 1 Synthetic route to the polymers. | |
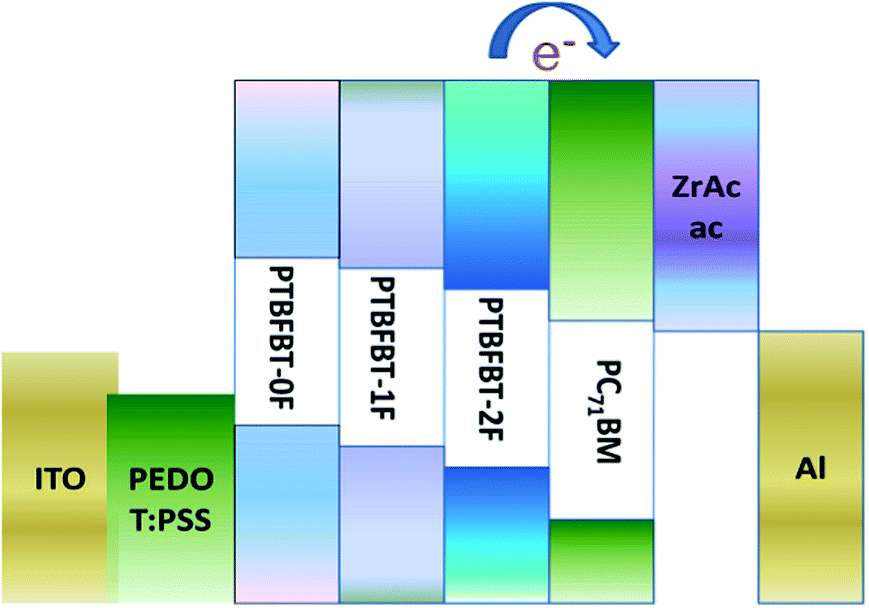
A solution-processable and stable cathode buffer layer (CBL) is also important for promoting PCE.29,30 The addition of a CBL is an effective strategy for simultaneously enhancing all device parameters.31,32 Tan et al. have demonstrated high performance PSCs by employing an as-prepared zirconium acetylacetonate film (ZrAcac) as a CBL.33 Therefore we tried to replace Ca by ZrAcac as a CBL and obtained an improved PCE from 4.2% to 5.0% for PTBFBT-2F.
Experimental section
Materials
2,6-Bis(trimethyltin)-4,8-bis(2′-ethylhexylthiophene)thieno[2,3-f]benzofuran (M1),27 4,7-bis(5-bromo-4-octylthiophen-2-yl)benzo[1,2,5]thiadiazole (M2),13 4,7-bis(5-bromo-4-octylthiophen-2-yl)-5-fluorobenzo[1,2,5]thiadiazole (M3),26 4,7-bis(5-bromo-4-octylthiophen-2-yl)-5,6-difluorobenzo[1,2,5]thiadiazole (M4)21 were synthesized according to the previously reported procedures. Palladium(0)tetrakis(triphenylphosphine) (Pd(PPh3)4) was purchased from Sigma-Aldrich Chemical Co., and it was used as received. Other reagents and solvents were used as received without further purification unless stated otherwise.
Characterization
1H NMR spectra of the polymers were recorded using an AVANCE III 500 MHz or AVANCE III 400 MHz spectrometer in chloroform-d solution at 298 K with tetramethylsilane (TMS) as internal reference (0 ppm). Number-average (Mn) and weight-average (Mw) molecular weights were measured by gel permeation chromatography (GPC) analysis with polystyrene as a standard and THF (HPLC grade) as the eluent at a flow rate of 1.0 mL min−1 at 35 °C. Thermogravimetric analysis (TGA) was performed on a Perkin-Elmer TGA-7 at a heating rate of 10 K min−1 under nitrogen atmosphere. UV-Vis absorption spectra were taken using a Shimadzu UV-2450 spectrophotometer. For solid state measurements, polymer solution in chloroform was cast on quartz plates. Optical bandgap was calculated from the onset of the absorption band. The electrochemical cyclic voltammetry was conducted on a Zahner IM6e Electrochemical Workstation, in a 0.1 mol L−1 acetonitrile (CH3CN) solution of tetrabutylammonium hexafluorophosphate (n-Bu4NPF6) at a scan rate of 100 mV s−1 with a glassy carbon working electrode, an Ag/AgCl reference electrode and a platinum wire counter electrode. Power X-ray diffraction (XRD) measurements were carried out with an 18 kW Rigaku X-ray diffraction system. The morphologies of the polymer/PC71BM blend films were investigated by tapping-mode with atomic force microscopy (AFM, Multimode 8 by Bruker). Transmission electron microscope (TEM) measurements were performed in a JEM-2100F.
Device fabrication
The PSCs were fabricated with a configuration of ITO/PEDOT:PSS (40 nm)/active layer/cathode. A thin layer of PEDOT:PSS (poly(3,4-ethylenedioxythiophene):poly(styrene-sulfonate)) was deposited through spin-coating on pre-cleaned ITO-coated glass with a PEDOT:PSS aqueous solution (Baytron PVP AI 4083 from H. C. Starck) at 3000 rpm and dried subsequently at 150 °C for 15 min in air, then the device was transferred to a nitrogen glove box, where the active blend layer of the polymer and fullerene derivative was spin-coated onto the PEDOT:PSS layer. For conjugated polymer: PC71BM PSCs, the active layer was formed by spin coating with ortho-dichlorobenzene (o-DCB) solution containing 10 mg mL−1 polymer. The ZrAcac CBL was simply prepared by spin-coating an ethanol solution (1 mg mL−1) on the photoactive layer at 3000 rpm for 30 s at room temperature, no thermal annealing or any other post-treatment was performed. Finally, the top electrode was deposited in a vacuum onto the active layer. The active area of the device was 5 mm2.
The measurements of J–V characteristics and EQE
Current density–voltage (J–V) characteristics of the devices were measured with a computer-controlled Keithley 236 Source Measure Unit. A xenon lamp coupled with AM 1.5G solar spectrum filters was used as the light source. The illumination intensity of 1000 W m−2 irradiation was calibrated using a standard monocrystal Si reference cell to ensure an exact light intensity. The external quantum efficiency (EQE) spectra were measured by a Stanford Research Systems model SR830 DSP lock-in amplifier coupled with WDG3 monochromator and 500 W xenon lamp.
Synthesis of the polymers
Synthesis of PTBFBT-0F. M1 (0.2 mmol, 178 mg) and M2 (0.2 mmol, 136 mg) were dissolved in toluene (10 mL) and DMF (2 mL) in a two-necked flask. The solution was flushed with argon (Ar) for 20 min, then Pd(PPh3)4 (15 mg) was added into the flask and this mixture was flushed with Ar for another 20 min. The reaction was heated to 110 °C gradually and stirred for 24 h at this temperature under Ar. Then the reaction mixture was cooled to room temperature and poured into methanol (100 mL) slowly. The resulting precipitate was filtered through a Soxhlet thimble, which was then subjected to Soxhlet extractions with methanol, hexane and chloroform. Finally the polymer was recovered as a solid from the chloroform fraction by rotary evaporation and a dark-blue solid was obtained (156 mg, 74%). GPC: Mn = 12.3 kDa, Mw = 30.0 kDa, PDI = 2.4. 1H NMR (400 MHz, CDCl3): δ 8.06 (br, 1H), 7.74 (br, 2H), 7.54 (br, 1H), 7.50 (br, 2H), 7.29 (br, 1H), 7.02 (br, 1H), 6.97 (br, 2H), 2.94 (br, 4H), 2.25 (br, 2H), 1.26–1.54 (br, 44H), 0.88–1.26 (br, 18H).
Synthesis of PTBFBT-1F. PTBFBT-1F was synthesized from M1 and M3 similarly to the synthesis of PTBFBT-0F. PTBFBT-1F was obtained as a blue solid (144 mg, 68%). GPC: Mn = 10.0 kDa, Mw = 20.9 kDa, PDI = 2.1. 1H NMR (500 MHz, CDCl3): δ 8.02 (br, 1H), 7.68 (br, 2H), 7.55 (br, 1H), 7.50 (br, 1H), 7.33 (br, 1H), 7.08 (br, 1H), 6.97 (br, 2H), 2.94 (br, 4H), 2.24 (br, 2H), 1.26–1.54 (44H, br), 0.88–1.26 (br, 18H).
Synthesis of PTBFBT-2F. PTBFBT-2F was synthesized from M1 and M4 similarly to the synthesis of PTBFBT-0F. PTBFBT-2F was obtained as a blue solid (142 mg, 63%). GPC: Mn = 12.9 kDa, Mw = 33.5 kDa, PDI = 2.6. 1H NMR (400 MHz, CDCl3): δ 8.06 (br, 1H), 7.75 (br, 2H), 7.53 (br, 1H), 7.30 (br, 1H), 7.04 (br, 1H), 6.99 (br, 2H), 2.93 (br, 4H), 2.23 (br, 2H), 1.26–1.54 (br, 44H), 0.88–1.26 (br, 18H).
Results and discussion
Synthesis and characterization
The general synthetic route to these polymers is sketched in Scheme 1. The target polymers PTBFBT-0F, PTBFBT-1F and PTBFBT-2F were obtained through Stille coupling reactions. The synthesized polymers were purified by continuous extractions with methanol, hexane and chloroform, and the chloroform fractions were recovered. All the polymers are highly soluble in common organic solvents such as chloroform, chlorobenzene and dichlorobenzene at room temperature. 1H NMR spectra of the polymers are shown in Fig. S1.† The molecular weights of the polymers were determined by GPC using THF as eluent and polystyrene as the standard. The Mw of PTBFBT-0F, PTBFBT-1F and PTBFBT-2F is 30.0 kDa, 20.9 kDa, 33.5 kDa with polydispersity indices (PDIs) of 2.4, 2.1 and 2.6, respectively. The related data are summarized in the Table 1.
Table 1 Polymerization results of PTBFBT-0F, PTBFBT-1F and PTBFBT-2F
| Polymer |
Mn (kDa) |
Mw (kDa) |
PDI |
| PTBFBT-0F |
12.3 |
30.0 |
2.4 |
| PTBFBT-1F |
10.0 |
20.9 |
2.1 |
| PTBFBT-2F |
12.9 |
33.5 |
2.6 |
Thermal stability
The availability of semiconducting polymers that can withstand exposure to elevated temperatures would bring a variety of new possibilities.34 The thermal properties of PTBFBT-0F, PTBFBT-1F and PTBFBT-2F were investigated by TGA under nitrogen atmosphere, and the results are presented in Fig. 1. As we can see, all of the resulting polymers exhibited excellent thermal stability with 5% weight loss at temperatures of 337 °C, 307 °C and 363 °C for PTBFBT-0F, PTBFBT-1F and PTBFBT-2F, respectively. This shows that the three polymers have excellent thermal stability for device fabrication.
 |
| | Fig. 1 TGA thermograms of these polymers with a heating rate of 10 K min−1 under nitrogen atmosphere. | |
X-ray diffraction analysis
XRD measurement has been used to investigate the crystalline properties of the conjugated polymers. Fig. S2† shows the XRD patterns of the three conjugated polymers. Obviously, no peak can be found in the region from 2 to 10 degrees, indicating that no ordered laminar packing can be formed in the three polymers. However, diffraction peaks around 25°, which correspond to the π–π stacking d-spacing of 3.56 Å, are broad and without prominent difference in intensity and location. This means that the fluorine number has little effect on the polymer chain packing structure.
Optical properties
The UV-Vis absorption spectra of these polymers in solution and as films are shown in Fig. 2, and the corresponding characteristics are summarized in Table 2. The absorption spectra of the three polymer films identically showed two major absorption peaks. One peak in the wavelength of ca. 400–500 nm is induced by the localized π–π* transition and the other is in the range of 500–700 nm originating from a typical ICT. The absorption spectra of the three polymers in films are broader and red-shifted relative to those in solution. This can be ascribed to the enhanced intermolecular interactions between the polymer main chains and the planarization effect of the π-conjugated polymer backbone. In the film state, PTBFBT-1F and PTBFBT-2F have similar onset (λonset) and maxima (λmax) values which were blue-shifted compared with those of PTBFBT-0F. This phenomenon can be explained by the strong electron-withdrawing nature of F increasing the electron density on the F atom. This leads to a permanent shift of π electrons which weakened the conjugation effect resulting in blue-shifted UV-Vis absorption. Introducing F on the polymer backbone makes the λonset and λmax of the polymers blue-shift which widened the energy bandgap,17,24,35,36 the corresponding data are summarized in Table 2.
 |
| | Fig. 2 UV-Vis absorption spectra of polymers in chloroform solution (a) and in thin films (b). | |
Table 2 Optical and electrochemical properties of these polymers
| Polymer |
Solution |
Film |
CV |
| λmax [nm] |
λmax [nm] |
λonset [nm] |
Eoptg [eV] |
φox/HOMO [V]/[eV] |
φred/LUMO [V]/[eV] |
Eecg [eV] |
| PTBFBT-0F |
601 |
624 |
754 |
1.64 |
0.57/−4.98 |
−1.27/−3.14 |
1.84 |
| PTBFBT-1F |
551 |
601 |
737 |
1.68 |
0.70/−5.11 |
−1.26/−3.15 |
1.96 |
| PTBFBT-2F |
589 |
601 |
730 |
1.69 |
0.72/−5.13 |
−1.08/−3.33 |
1.80 |
Electrochemical properties
To determine the HOMO and lowest unoccupied molecular orbital (LUMO) levels of these polymers, cyclic voltammetry (CV) measurements were carried out under nitrogen in a three-electrode cell using 0.1 M n-Bu4NPF6 in anhydrous CH3CN as the supporting electrolyte. The CV data and curves of polymers are shown in Table 2 and Fig. 3. The corresponding energy-band diagrams are shown in Fig. 4. HOMO and LUMO levels were determined using the equation: EHOMO = −e(Eox + 4.41) and ELUMO = −e(Ere + 4.41).37 The onset of oxidation and reduction potential of PTBFBT-0F were observed at 0.57 V and −1.27 V, corresponding to HOMO and LUMO levels at −4.98 eV and −3.14 eV. For PTBFBT-1F, the onsets were observed at 0.70 V and −1.26 V, corresponding to the HOMO and LUMO levels at −5.11 eV and −3.15 eV. For PTBFBT-2F, the onsets were observed at 0.72 V and −1.08 V, corresponding to the HOMO and LUMO levels at −5.13 eV and −3.33 eV. The electron-withdrawing nature of the F atom lowers the HOMO energy level of the fluorinated polymer compared with that of the non-fluorinated analog. Muhammet E. Köse et al. further proved it by theoretical calculations.38 There is a minor difference in the HOMO energy level between PTBFBT-1F and PTBFBT-2F. Thus, the Voc of PTBFBT-1F and PTBFBT-2F are similar but higher than that of PTBFBT-0F.
 |
| | Fig. 3 Cyclic voltammograms of three polymers. | |
 |
| | Fig. 4 Energy-band diagram of the materials involved in PSCs. | |
Photovoltaic properties
Photovoltaic properties of the three polymers were investigated in BHJ solar cells with the conventional device configuration of glass/ITO/PEDOT:PSS/polymer:PC71BM/Ca/Al. The blend ratios (w/w) of polymer and PC71BM were changed from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:3. For all polymers, devices with a D:A weight ratio of 1:1 showed the best performance of 3.0%, 3.6% and 4.2% for PTBFBT-0F, PTBFBT-1F and PTBFBT-2F, respectively. Fig. 5 shows typical J–V curves, measured under AM 1.5 illumination, 100 mW cm−2. The representative characteristics of the solar cells are summarized in Table 3. A stepwise increase in FF was observed with increasing number of fluorine atoms. The insertion of one fluorine atom into the BT moiety causes the FF to increase by 7.5% to 46%. Upon introduction of the second fluorine atom, the FF was further improved by 16.2% to 53% for PTBFBT-2F. This was probably due to the noncovalent attractive interactions between S (in thiophene) and F, and between C–H (in thiophene) and N (in BT) which minimize the torsional angle (Fig. S3†).39 The smaller torsional angle will maximize the planarity of the polymer chain which can result in an efficient charge transport. Noncovalent coulomb interactions have been utilized to increase the planarity and ordering of polymer chains by many groups.40–42 Besides the FF, the Voc was improved for PTBFBT-1F and PTBFBT-2F when compared with PTBFBT-0F due to the lower HOMO level. However, PTBFBT-1F and PTBFBT-2F exhibited a slightly lower Jsc probably due to blue shifted absorption. Nonetheless, it requires a balance between Voc, FF and Jsc to reach optimum efficiencies. Therefore, PTBFBT-0F has the lowest PCE resulting from having the lowest Voc and FF although it has the highest Jsc. While PTBFBT-2F-based solar cells showed elevated PCE up to 4.2% due to the large increase in FF. These results indicate that the photovoltaic properties of conjugated polymer can be effectively tuned by fluorination.18,43 The correlation between the values of Voc, Jsc, FF, PCE, hole and electron mobilities for the three polymers are shown in Fig. S4,† and related data are summarized in Table 3.
:1 to 1:3. For all polymers, devices with a D:A weight ratio of 1:1 showed the best performance of 3.0%, 3.6% and 4.2% for PTBFBT-0F, PTBFBT-1F and PTBFBT-2F, respectively. Fig. 5 shows typical J–V curves, measured under AM 1.5 illumination, 100 mW cm−2. The representative characteristics of the solar cells are summarized in Table 3. A stepwise increase in FF was observed with increasing number of fluorine atoms. The insertion of one fluorine atom into the BT moiety causes the FF to increase by 7.5% to 46%. Upon introduction of the second fluorine atom, the FF was further improved by 16.2% to 53% for PTBFBT-2F. This was probably due to the noncovalent attractive interactions between S (in thiophene) and F, and between C–H (in thiophene) and N (in BT) which minimize the torsional angle (Fig. S3†).39 The smaller torsional angle will maximize the planarity of the polymer chain which can result in an efficient charge transport. Noncovalent coulomb interactions have been utilized to increase the planarity and ordering of polymer chains by many groups.40–42 Besides the FF, the Voc was improved for PTBFBT-1F and PTBFBT-2F when compared with PTBFBT-0F due to the lower HOMO level. However, PTBFBT-1F and PTBFBT-2F exhibited a slightly lower Jsc probably due to blue shifted absorption. Nonetheless, it requires a balance between Voc, FF and Jsc to reach optimum efficiencies. Therefore, PTBFBT-0F has the lowest PCE resulting from having the lowest Voc and FF although it has the highest Jsc. While PTBFBT-2F-based solar cells showed elevated PCE up to 4.2% due to the large increase in FF. These results indicate that the photovoltaic properties of conjugated polymer can be effectively tuned by fluorination.18,43 The correlation between the values of Voc, Jsc, FF, PCE, hole and electron mobilities for the three polymers are shown in Fig. S4,† and related data are summarized in Table 3.
 |
| | Fig. 5 J–V (a) and EQE (b) curves of polymer/PC71BM-based regular single solar cells under AM 1.5G illumination, 100 mW cm−2. aWith Ca, bwith ZrAcac. | |
Table 3 Device parameters for polymer/PC71BM-based PSCs
| Active layer polymer:PC71BM = 1:1 |
CBL |
Voc (V) |
Jsc (mA cm−2) |
PCE (%) |
FF (%) |
Hole mobilities (cm2 V−1 s−1) |
Electron mobilities (cm2 V−1 s−1) |
| PTBFBT-0F |
Ca |
0.63 |
11.18 |
3.0 |
42 |
2.65 × 10−6 |
2.25 × 10−5 |
| PTBFBT-0F |
ZrAcac |
0.65 |
12.37 |
4.1 |
49 |
| PTBFBT-1F |
Ca |
0.77 |
10.28 |
3.6 |
46 |
1.79 × 10−6 |
7.15 × 10−7 |
| PTBFBT-1F |
ZrAcac |
0.77 |
10.76 |
4.2 |
50 |
| PTBFBT-2F |
Ca |
0.75 |
10.54 |
4.2 |
53 |
1.52 × 10−5 |
1.83 × 10−5 |
| PTBFBT-2F |
ZrAcac |
0.74 |
13.32 |
5.0 |
50 |
In order to further optimize the performance of three polymers, we use ZrAcac as a cathode interlayer between the active layer and Al electrode which induces an increase of Jsc values, resulting in a further increase of PCE from 3.0% to 4.1% for PTBFBT-0F, 3.6% to 4.2% for PTBFBT-1F, 4.2% to 5.0% for PTBFBT-2F. All the Jsc were in good agreement with the EQE spectra (shown in Fig. 5) within an experimental error of 8%.
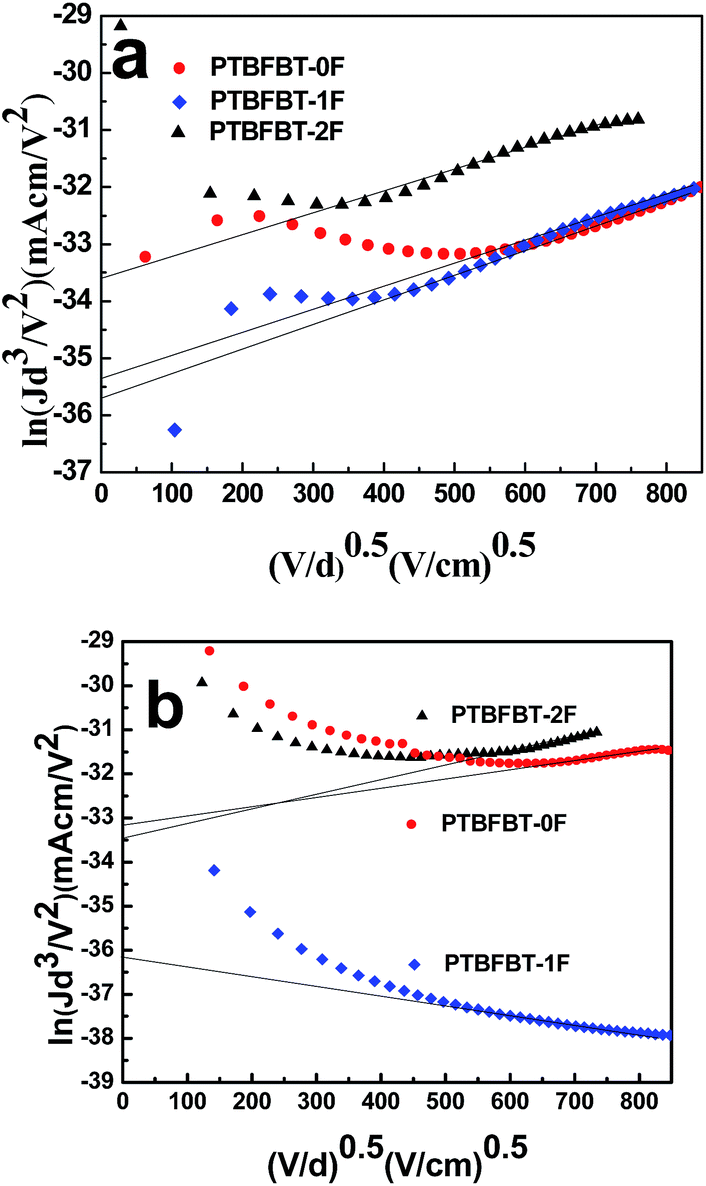
Charge transport
In order to quantify charge mobilities, a hole-only device (ITO/PEDOT:PSS/polymer:PC71BM/Au) and an electron-only device (Al/polymer:PC71BM/Al) were prepared using optimized BHJ films and their J–V characteristics were analyzed by the space charge limited current (SCLC) method. The hole and electron mobilities are calculated by the Poole–Frenkel law.44 Herein, J is current density, d stands for the thickness of the device, and V = Vappl − Vbi, where Vappl is the applied potential and Vbi is the built-in potential. The SCLC model can be described by the equation:
The results are plotted as ln(Jd3/V2) vs. (V/d)0.5 as shown in Fig. 6. The hole and electron mobilities of PTBFBT-0F, PTBFBT-1F, PTBFBT-2F were calculated to be 2.65 × 10−6 cm2 V−1 s−1 and 2.25 × 10−5 cm2 V−1 s−1, 1.79 × 10−6 cm2 V−1 s−1 and 7.15 × 10−7 cm2 V−1 s−1, 1.52 × 10−5 cm2 V−1 s−1 and 1.83 × 10−5 cm2 V−1 s−1, respectively. The relatively higher and balanced charge transport of PTBFBT-2F leads to a high FF up to 53%.45
 |
| | Fig. 6 ln(Jd3/V2)–(V/d)0.5 plots for the polymer:PC71BM (1:1, w/w) based hole-only device (a) and electron-only device (b). | |
Morphology analysis
The morphology of the BHJ layer is very important for device performance. The donor plays a major role in light absorption and exciton generation. The acceptor splits the excitons and creates electron pathways. Ideally, donors and acceptors would form two distinct continuous phases where the average size of the domains will be tens of nanometers in size.46
We investigated the morphologies of polymer and PC71BM blends spin-coated from their o-DCB solutions using AFM. Surface topography and phase images were taken for each film and are shown in Fig. S5.† Root-mean-square (RMS) roughness increases from 0.66 nm for the non-fluorinated case, 1.05 nm for mono-fluorinated to 1.88 nm for di-fluorinated.47 The nanoscale morphology of blends of three polymers with PC71BM showed bicontinuous networks and are quite similar with only small variations of the RMS.
Another powerful technique is transmission electron microscopy (TEM). The specific density difference between polymer and PCBM enables the polymer-rich and fullerene-rich regions to be mapped. In the PTBFBT-0F/PC71BM blend film images (Fig. 7a), the brighter areas are attributed to the polymer-rich regions, here interconnected via a network of PCBM-rich boundaries.48 The slightly coarse phase-separated morphology, compared with PTBFBT-2F/PC71BM blend film, is likely to limit the efficiency of exciton diffusion to the polymer/PCBM interfaces.49,50 However, it could be observed that the PTBFBT-2F/PC71BM exhibits moderate homogeneity and there was no detrimental phase segregation and good miscibility with PC71BM which can explain the excellent FF (Fig. 7c).
 |
| | Fig. 7 TEM images of PTBFBT-0F/PC71BM film (a), PTBFBT-1F/PC71BM film (b), PTBFBT-2F/PC71BM film (c). | |
Conclusion
In summary, we have synthesized three copolymers from electron-deficient BT with different numbers of fluorine atoms as the acceptor unit and TBF as the donor unit, named PTBFBT-0F, PTBFBT-1F and PTBFBT-2F. These polymers were thoroughly investigated. All these polymers display good solution-processed properties, sufficient thermal stability and low bandgap. A decrease in the HOMO level and a slightly blue-shifted absorption of the fluorinated polymer relative to the non-fluorinated polymer were observed. PTBFBT-1F and PTBFBT-2F have similar performance in photophysics and photovoltaic properties except for FF. The relatively high and balanced hole and electron transport mobility, bicontinuous and homogeneous morphology led to a high FF of PTBFBT-2F. Preliminary photovoltaic cells based on PTBFBT-0F, PTBFBT-1F, PTBFBT-2F as the donor showed increasing efficiency of 3.0%, 3.6%, 4.2%, respectively. Furthermore, 5.0% efficiency with PTBFBT-2F as the donor was obtained when using ZrAcac as CBL. Therefore, introducing different numbers of fluorine atoms into the acceptor units in D–A polymers is be a promising method to effectively tune their photophysics and photovoltaic properties.
Acknowledgements
This work was supported by NSFC (nos 51173206, 21161160443). The authors acknowledge the NMR measurements from the Modern Analysis and Testing Center of CSU.
Notes and references
- G. Yu, J. Gao, J. Hummelen, F. Wudl and A. Heeger, Science, 1995, 270, 1789–1790 CAS.
- C. C. Chen, W. H. Chang, K. Yoshimura, K. Ohya, J. You, J. Gao, Z. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- C. C. Chen, W. H. Chang, K. Yoshimura, K. Ohya, J. B. You, J. Gao, Z. R. Hong and Y. Yang, Adv. Mater., 2014, 26, 5670–5677 CrossRef CAS PubMed.
- J. B. You, L. T. Dou, K. Yoshimura, T. Kato, K. Ohya, T. Moriarty, K. Emery, C. C. Chen, J. Gao, G. Li and Y. Yang, Nat. Commun., 2013, 4, 1446 CrossRef PubMed.
- J. B. You, C. C. Chen, Z. R. Hong, K. Yoshimura, K. Ohya, R. Xu, S. L. Ye, J. Gao, G. Li and Y. Yang, Adv. Mater., 2013, 25, 3973–3978 CrossRef CAS PubMed.
- Y. Liu, J. Zhao, Z. Li, C. Mu, W. Ma, H. Hu, K. Jiang, H. Lin, H. Ade and H. Yan, Nat. Commun., 2014, 5, 30 Search PubMed.
- W. W. Li, A. Furlan, K. H. Hendriks, M. M. Wienk and R. A. J. Janssen, J. Am. Chem. Soc., 2013, 135, 5529–5532 CrossRef CAS PubMed.
- S. Schumann, R. Da Campo, B. Illy, A. C. Cruickshank, M. A. McLachlan, M. P. Ryan, D. J. Riley, D. W. McComb and T. S. Jones, J. Mater. Chem., 2011, 21, 2381–2386 RSC.
- V. Kumar, H. M. Wang and C. Rodenburg, Org. Electron., 2014, 15, 2059–2067 CrossRef CAS PubMed.
- K. Zhang, C. M. Zhong, S. J. Liu, C. Mu, Z. K. Li, H. Yan, F. Huang and Y. Cao, ACS Appl. Mater. Interfaces, 2014, 6, 10429–10435 CAS.
- J. W. Shim, C. Fuentes-Hernandez, Y. H. Zhou, A. Dindar, T. M. Khan, A. J. Giordano, H. Cheun, M. Yun, S. R. Marder and B. Kippelen, Adv. Energy Mater., 2014, 4, 1400048 Search PubMed.
- A. B. Yusoff, S. J. Lee, J. Kim, F. K. Shneider, W. J. da Silva and J. Jang, ACS Appl. Mater. Interfaces, 2014, 6, 13079–13087 Search PubMed.
- C. Kitamura, S. Tanaka and Y. Yamashita, Chem. Mater., 1996, 8, 570–578 CrossRef CAS.
- A. C. Stuart, J. R. Tumbleston, H. X. Zhou, W. T. Li, S. B. Liu, H. Ade and W. You, J. Am. Chem. Soc., 2013, 135, 1806–1815 CrossRef CAS PubMed.
- T. Okamoto, K. Nakahara, A. Saeki, S. Seki, J. H. Oh, H. B. Akkerman, Z. Bao and Y. Matsuo, Chem. Mater., 2011, 23, 1646–1649 CrossRef CAS.
- J. W. Jo, J. W. Jung, H.-W. Wang, P. Kim, T. P. Russell and W. H. Jo, Chem. Mater., 2014, 26, 4214–4220 CrossRef CAS.
- H. J. Son, W. Wang, T. Xu, Y. Y. Liang, Y. E. Wu, G. Li and L. P. Yu, J. Am. Chem. Soc., 2011, 133, 1885–1894 CrossRef CAS PubMed.
- M. Zhang, X. Guo, S. Zhang and J. Hou, Adv. Mater., 2014, 26, 1118–1123 CrossRef CAS PubMed.
- B. Carsten, J. M. Szarko, H. J. Son, W. Wang, L. Y. Lu, F. He, B. S. Rolczynski, S. J. Lou, L. X. Chen and L. P. Yu, J. Am. Chem. Soc., 2011, 133, 20468–20475 CrossRef CAS PubMed.
- B. S. Rolczynski, J. M. Szarko, H. J. Son, Y. Y. Liang, L. P. Yu and L. X. Chen, J. Am. Chem. Soc., 2012, 134, 4142–4152 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- H. Bronstein, J. M. Frost, A. Hadipour, Y. Kim, C. B. Nielsen, R. S. Ashraf, B. P. Rand, S. Watkins and I. McCulloch, Chem. Mater., 2013, 25, 277–285 CrossRef CAS.
- B. C. Schroeder, Z. Huang, R. S. Ashraf, J. Smith, P. D’Angelo, S. E. Watkins, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Funct. Mater., 2012, 22, 1663–1670 CrossRef CAS.
- Y. Zhang, S. C. Chien, K. S. Chen, H. L. Yip, Y. Sun, J. A. Davies, F. C. Chen and A. K. Jen, Chem. Commun., 2011, 47, 11026–11028 RSC.
- B. Liu, Y. Zou, S. Ye, Y. He and K. Zhou, RSC Adv., 2011, 1, 424–428 RSC.
- L. Xiao, B. Liu, X. Chen, Y. Li, W. Tang and Y. Zou, RSC Adv., 2013, 3, 11869 RSC.
- L. Fan, R. Cui, X. Guo, D. Qian, B. Qiu, J. Yuan, Y. Li, W. Huang, J. Yang, W. Liu, X. Xu, L. Li and Y. Zou, J. Mater. Chem. C, 2014, 2, 5651 RSC.
- Y. Huang, X. Guo, F. Liu, L. Huo, Y. Chen, T. P. Russell, C. C. Han, Y. Li and J. Hou, Adv. Mater., 2012, 24, 3383–3389 CrossRef CAS PubMed.
- G. Li, C. W. Chu, V. Shrotriya, J. Huang and Y. Yang, Appl. Phys. Lett., 2006, 88, 253503 CrossRef PubMed.
- H.-H. Liao, L.-M. Chen, Z. Xu, G. Li and Y. Yang, Appl. Phys. Lett., 2008, 92, 173303 CrossRef PubMed.
- M. Lv, S. Li, J. J. Jasieniak, J. Hou, J. Zhu, Z. Tan, S. E. Watkins, Y. Li and X. Chen, Adv. Mater., 2013, 25, 6889–6894 CrossRef CAS PubMed.
- Z.-G. Zhang, B. Qi, Z. Jin, D. Chi, Z. Qi, Y. Li and J. Wang, Energy Environ. Sci., 2014, 7, 1966 CAS.
- Z. Tan, S. Li, F. Wang, D. Qian, J. Lin, J. Hou and Y. Li, Sci. Rep., 2014, 4, 4691 Search PubMed.
- S. Cho, J. H. Seo, S. H. Park, S. Beaupre, M. Leclerc and A. J. Heeger, Adv. Mater., 2010, 22, 1253–1257 CrossRef CAS PubMed.
- Y. Sakamoto, S. Komatsu and T. Suzuki, J. Am. Chem. Soc., 2001, 123, 4643–4644 CrossRef CAS.
- J. Kim, M. H. Yun, G.-H. Kim, J. Lee, S. M. Lee, S.-J. Ko, Y. Kim, G. K. Dutta, M. Moon and S. Y. Park, ACS Appl. Mater. Interfaces, 2014, 6, 7523–7534 CAS.
- B. Liu, X. Chen, Y. Zou, Y. He, L. Xiao, X. Xu, L. Li and Y. Li, Polym. Chem., 2013, 4, 470–476 RSC.
- A. Iyer, J. Bjorgaard, T. Anderson and M. E. Köse, Macromolecules, 2012, 45, 6380–6389 CrossRef CAS.
- T. L. Nguyen, H. Choi, S. J. Ko, M. A. Uddin, B. Walker, S. Yum, J. E. Jeong, M. H. Yun, T. J. Shin, S. Hwang, J. Y. Kim and H. Y. Woo, Energy Environ. Sci., 2014, 7, 3040 CAS.
- Y. F. Wang, S. R. Parkin, J. Gierschner and M. D. Watson, Org. Lett., 2008, 10, 3307–3310 CrossRef CAS PubMed.
- X. Guo, J. Quinn, Z. Chen, H. Usta, Y. Zheng, Y. Xia, J. W. Hennek, R. P. Ortiz, T. J. Marks and A. Facchetti, J. Am. Chem. Soc., 2013, 135, 1986–1996 CrossRef CAS PubMed.
- X. G. Guo, F. S. Kim, S. A. Jenekhe and M. D. Watson, J. Am. Chem. Soc., 2009, 131, 7206–7207 CrossRef CAS PubMed.
- L. Yang, J. R. Tumbleston, H. Zhou, H. Ade and W. You, Energy Environ. Sci., 2013, 6, 316 CAS.
- W. Pasveer, J. Cottaar, C. Tanase, R. Coehoorn, P. Bobbert, P. Blom, D. De Leeuw and M. Michels, Phys. Rev. Lett., 2005, 94, 206601 CrossRef CAS.
- Y. Lin, J. Wang, S. Dai, Y. Li, D. Zhu and X. Zhan, Adv. Energy Mater., 2014, 4, 1400420 Search PubMed.
- F. Liu, Y. Gu, J. W. Jung, W. H. Jo and T. P. Russell, J. Polym. Sci., Part B: Polym. Phys., 2012, 50, 1018–1044 CrossRef CAS.
- W. Wang, W. Xu, L. Cosimbescu, D. Choi, L. Li and Z. Yang, Chem. Commun., 2012, 48, 6669–6671 RSC.
- J. Warnan, A. El Labban, C. Cabanetos, E. T. Hoke, P. K. Shukla, C. Risko, J.-L. Brédas, M. D. McGehee and P. M. Beaujuge, Chem. Mater., 2014, 26, 2299–2306 CrossRef CAS.
- B. A. Collins, Z. Li, J. R. Tumbleston, E. Gann, C. R. McNeill and H. Ade, Adv. Energy Mater., 2013, 3, 65–74 CrossRef CAS.
- S. J. Lou, J. M. Szarko, T. Xu, L. Yu, T. J. Marks and L. X. Chen, J. Am. Chem. Soc., 2011, 133, 20661–20663 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1 showing 1H NMR spectra of the polymers, Fig. S2 showing the X-ray diffraction patterns of three polymers, Fig. S3 showing noncovalent attractive interactions within the polymer chain. Fig. S4 showing AFM images (3 μm × 3 μm). See DOI: 10.1039/c5ra03405a |
| ‡ R. Cui and L. Fan contributed to this work equally. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.