
Synthesis and electrochemical characteristics of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) cathode materials for lithium ion batteries
Chao Zhang,
Peiyu Hou,
Xixi Shi*,
Dawei Song,
Jishun Song and
Lianqi Zhang*
School of Materials Science and Engineering, Tianjin University of Technology, Tianjin 300384, China. E-mail: tianjinzhanglq@163.com; cshi6@163.com; Fax: +86 22 60214028; Tel: +86 22 60214577
First published on 14th April 2015
Abstract
According to the tetrahedral phase diagram of LiNiO2–LiCoO2–LiMnO2–Li2MnO3, a series of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) have been designed to explore new Li-rich solid solution cathode materials. The effects of Li1.2Ni0.2Mn0.6O2, Li1.2Co0.4Mn0.4O2 and Li1.2Ni0.4Mn0.4O2 content in solid solutions on structure and electrochemical properties are investigated. Micro-sized spherical or ellipsoidal precursors are first prepared via a carbonate co-precipitation route. After calcination with lithium sources, all samples are indexed to a typical layered structure with an R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group as detected by X-ray diffraction (XRD). It is found that the introduction of Co can improve the tap density. However, these Co referred samples reveal lower discharge specific capacities and inferior cycle life. For these Co-free materials with high Ni content, for instance Li1.2Ni0.3Mn0.5O2, although low capacity is observed in the initial cycle, a large capacity of above 250 mA h g−1 is achieved after about 10 cycles. Importantly, the activated Li1.2Ni0.3Mn0.5O2 material still delivers a high capacity of over 230 mA h g−1 after 70 cycles, displaying superior cycle stability. These results may be instructive in designing and exploring high performance cathode materials for advanced LIBs.
m space group as detected by X-ray diffraction (XRD). It is found that the introduction of Co can improve the tap density. However, these Co referred samples reveal lower discharge specific capacities and inferior cycle life. For these Co-free materials with high Ni content, for instance Li1.2Ni0.3Mn0.5O2, although low capacity is observed in the initial cycle, a large capacity of above 250 mA h g−1 is achieved after about 10 cycles. Importantly, the activated Li1.2Ni0.3Mn0.5O2 material still delivers a high capacity of over 230 mA h g−1 after 70 cycles, displaying superior cycle stability. These results may be instructive in designing and exploring high performance cathode materials for advanced LIBs.
1 Introduction
The depletion of traditional energy resources as well as the desire to reduce high CO2 emissions associated with energy production indicate that clean energy storage is now becoming more important than ever. Rechargeable lithium-ion batteries (LIBs) have become one of the key points of the development of clean energy, and their application range is continually expanded. Since Sony announced the commercial availability of rechargeable LIBs, layered LiMO2 has been widely used as cathode materials.1,2Recently, much attention has focused on Li-rich layered materials as well as their related composite materials with spinel phases as cathode material for the next generation LIBs.3–31 Li-rich layered materials usually show interesting electrochemical properties, such as large reversible capacity of over 250 mA h g−1 and excellent cycle life when cycled to an upper cutoff voltage of about 4.8 V. These materials can be generally expressed as formula Li1+zM1−zO2 (M can be one or multi-metal ions, z > 0).25 In our opinion, Li-rich layered ordered/disordered materials Li1+zM1−zO2 can be all regarded as the LiAO2–Li2BO3 solid solutions, which are based on the structure of rock salt; layered ordered structure can be further regarded as a solid solution material between layered LiAO2 and Li2BO3.25 Considering that the complexity in material composition and structure as well as valence variability is extremely easy to cause confusion on the reader to understand, we have proposed a tetrahedral phase diagram of LiNiO2–LiCoO2–LiMnO2–Li2MnO3, as shown in Fig. 1, to describe and understand the nature of such materials.24,25
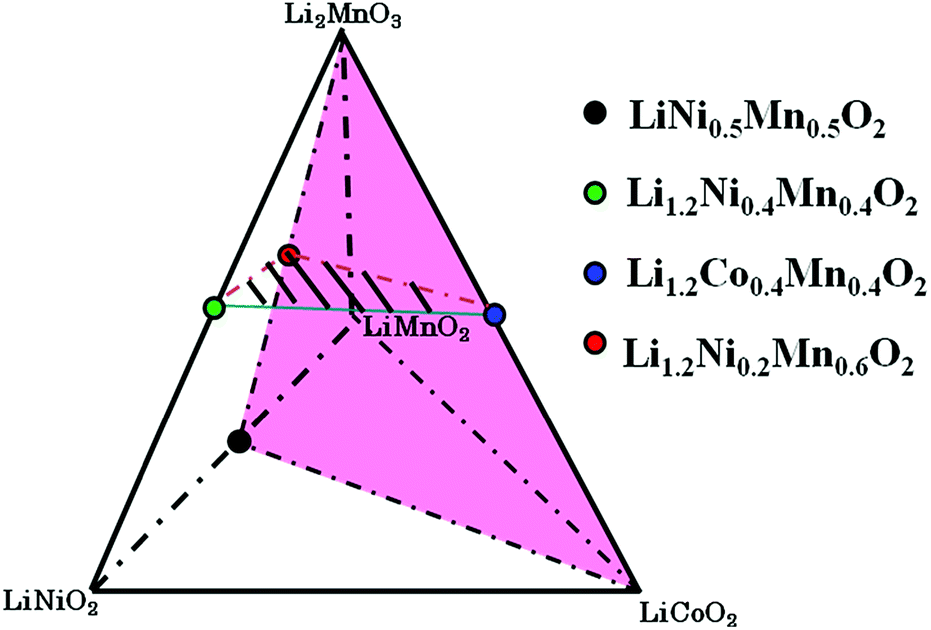 | ||
| Fig. 1 The tetrahedral phase diagram of LiNiO2–LiCoO2–LiMnO2–Li2MnO3. | ||
According to the previous reports,5,7,8,13,26–28 it may be deduced that pure phased solid solutions can be formed in the space of LiNiO2–LiCoO2–LiNi0.5Mn0.5O2–Li2MnO3 via a high temperature solid reaction.25 In this work, in order to further support the point as well as explore new Li-rich solid solution materials, the aim of this paper is to synthesize a series of Li-rich layered materials with the nominal compositions of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1), as better described in Fig. 2, and investigate the effects of Li1.2Ni0.2Mn0.6O2, Li1.2Co0.4Mn0.4O2 and Li1.2Ni0.4Mn0.4O2 content in solid solutions on structure and electrochemical properties. Theoretically, such a substitution can be assumed on line 1 that one Ni3+ replaces one Mn4+, accompanying oxidization of one Ni2+ to Ni3+ with decrease of x value. Similarly, on line 2 there exists a substitution equation (valence of 2Co3+ is equal to that of Ni2+ and Mn4+, Ni2+ + Mn4+ = 2Co3+), and on line 3 Ni3+ is gradually substituted for Co3+ with the increase of y value. In addition, a carbonate co-precipitation route followed by a heat-treatment reaction is used to prepare monodisperse spherical particles.
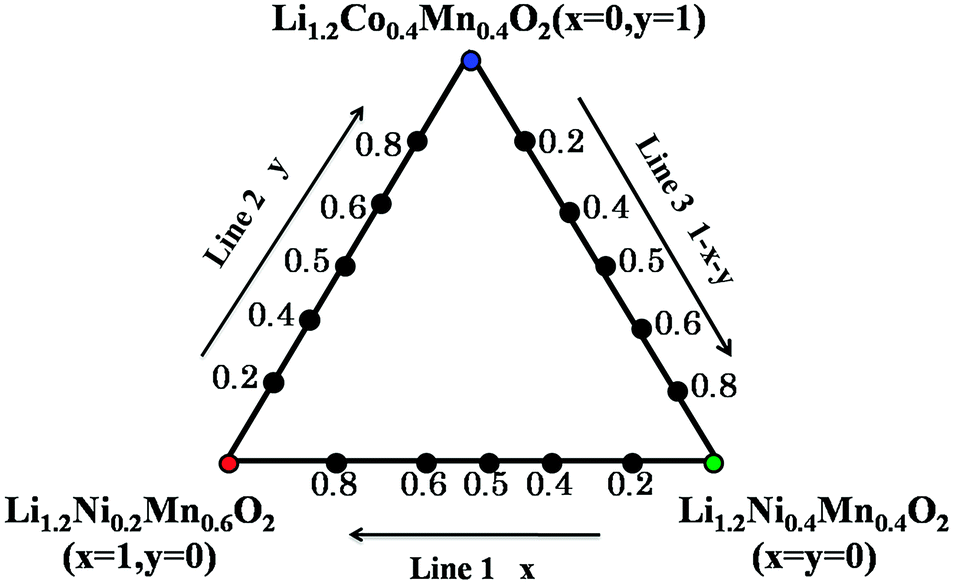 | ||
| Fig. 2 The triangle phase diagram of Li1.2Ni0.2Mn0.6O2–Li1.2Co0.4Mn0.4O2–Li1.2Ni0.4Mn0.4O2. Any compositions in the diagram can be all express with x and y values, that is Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1). | ||
2 Experimental
To synthesize the (Ni0.25Mn0.75)x(Co0.5Mn0.5)y(Ni0.5Mn0.5)1−x−yCO3 (0 ≤ x + y ≤ 1) carbonate precursors, stoichiometric NiSO4·6H2O, CoSO4·7H2O and MnSO4·H2O were dissolved with a concentration of 2.0 mol L−1 as the starting materials. A mixed aqueous solution with 2.0 mol L−1 Na2CO3 and 0.2 mol L−1 NH4OH (as the chelating agent) was also prepared as the starting alkali solution. The starting alkali and sulfates solutions were simultaneously pumped into a continuously stirred tank reactor (CSTR, capacity of 10 L) with 4 L water under a nitrogen atmosphere and reacted by controlling pH value as 8.5, leading to the (Ni0.25Mn0.75)x(Co0.5Mn0.5)y(Ni0.5Mn0.5)1−x−yCO3.The obtained precursor powders were separated from the aqueous medium. After washed and filtered several times, they were dried inside a drying oven at 100 °C for 24 hours. Thereafter, stoichiometric amounts of Li2CO3 were well mixed with the carbonate precursors. The mixture was firstly calcined at 600 °C for 10 h to decompose the carbonate precursors and then calcined at 900 °C for 15 h to obtain the compounds.
Precursors were periodically collected during the co-precipitation experiment and the particle size distribution was measured by a particle size analyzer (OMEC, LS-POP(6), China). X-ray diffractometry (XRD, Rigaku D/MAX-2500 Japan) was employed to characterize structure of the prepared materials. XRD data were obtained at 2θ = 10°–80° with a step size of 0.02°, using Cu Kα radiation. The morphology of synthesized materials was observed by a Scanning Electron Microscope (SEM, JMS-6700F, JEOL, Japan). The tap density of materials was tested by ZS Tap Density (ZS-201).
For fabrication of cathode electrodes, the prepared materials were mixed with acetylene black and PVDF (80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in weight) in NMP. The obtained slurry was coated onto Al foil and dried at 80 °C for a day, followed by a roll-pressing. Prior to use, the electrodes were dried again at 120 °C for half a day in a vacuum oven. The electrodes were electrochemically characterized using a CR2032 type of coin cell with lithium foil as the anode and 1 M LiPF6 in ethylene carbonate and diethyl carbonate (1:1 in volume) as the electrolyte. The cells were preliminarily charged and discharged in the voltage range of 2.0–4.8 V (versus Li) at 25 °C at a constant current density of 20 mA g−1.
:10:10 in weight) in NMP. The obtained slurry was coated onto Al foil and dried at 80 °C for a day, followed by a roll-pressing. Prior to use, the electrodes were dried again at 120 °C for half a day in a vacuum oven. The electrodes were electrochemically characterized using a CR2032 type of coin cell with lithium foil as the anode and 1 M LiPF6 in ethylene carbonate and diethyl carbonate (1:1 in volume) as the electrolyte. The cells were preliminarily charged and discharged in the voltage range of 2.0–4.8 V (versus Li) at 25 °C at a constant current density of 20 mA g−1.
3 Results and discussion
XRD patterns of as-prepared carbonate precursors (Ni0.25Mn0.75)x(Co0.5Mn0.5)y(Ni0.5Mn0.5)1−x−yCO3 (0 ≤ x + y ≤ 1) are revealed in Fig. 3. It can be seen that the precipitates are carbonate, because all reflections can be indexed to a space group of Rc.29–31 The regular shift in 2θ position can be found with careful observation, which can be clearly seen especially from Fig. 3(b′) and (c′). These regular changes probably can result from the variation of transitional metals ion radius in (Ni0.25Mn0.75)x(Co0.5Mn0.5)y(Ni0.5Mn0.5)1−x−yCO3. A similar discussion will be done in detail in XRD data of lithiated compounds in Fig. 6. Therefore, the aimed carbonate precursors are successfully synthesized via the co-precipitation method in this work.
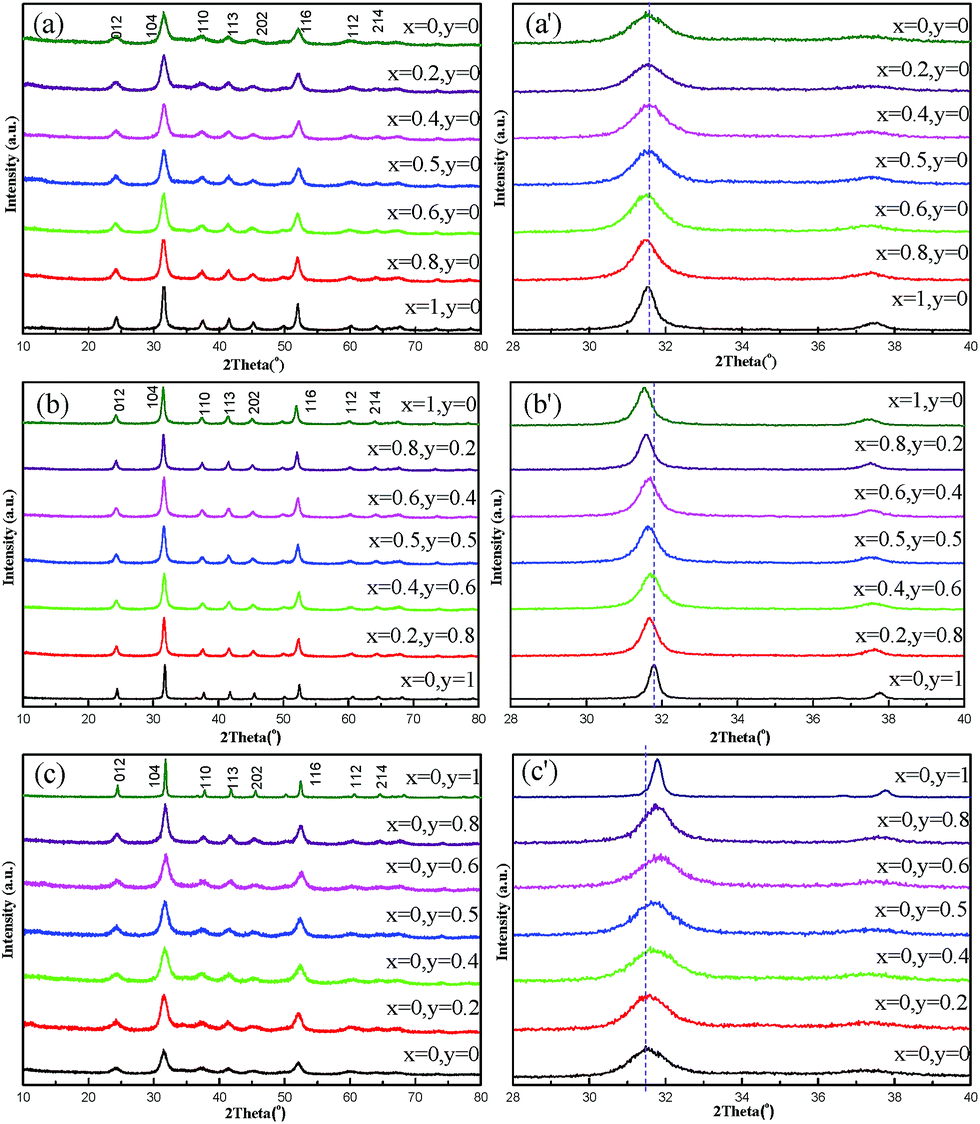 | ||
| Fig. 3 XRD patterns of as-prepared carbonate precursors via a co-precipitation route. Full XRD patterns of samples on (a) line 1, (b) line 2 and (c) line 3, respectively. Local magnified XRD patterns near the (104) peak on (a′) line 1, (b′) line 2 and (c′) line 3. | ||
From the typical SEM images of carbonate precursors on line 1 (Fig. 4), all precursors have spherical or ellipsoidal secondary particles aggregated from nano-scaled primary particles. It is obvious that the smooth surface of the powders is achieved, meaning that primary particles are closely packed during co-precipitation. All samples have monodisperse particles, suggesting excellent flowability of material. The particle size distributions of carbonate precursors are analyzed and the results are shown in Fig. 5. Clearly, all precursors have a single and normal distribution, but particle size seems to depend somewhat on the component.29–31 We believe that the nucleus formation and crystal growth highly depends on the reaction conditions in CSRT and the ratio of Ni/Co/Mn. In our work, the high Ni element appears to hinder the growth of precursors, which is probably attributed to the lower Ksp of NiCO3 compared with that of CoCO3 and MnCO3.
 | ||
| Fig. 4 The typical SEM images of as-prepared carbonate precursors: samples on line 1. | ||
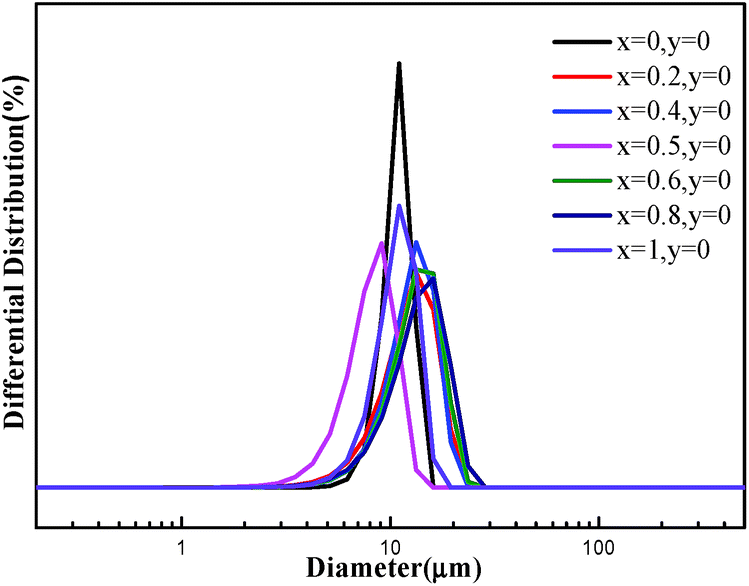 | ||
| Fig. 5 Particle size distribution curves of as-prepared carbonate precursors on line 1. | ||
The micro-spherical powders have close packing and monodisperse performances, and thus high tap density can be anticipated for as-prepared precursors. The detailed tap densities are listed in Table 1, in which the tap densities of Ni0.25+xMn0.75−xCO3 (0 ≤ x ≤ 0.2) compositions on line 1 remain around 1.60 g cm−3 even increasing Ni content. Interestingly, an increased tap density presents as Co content ranges from 0 to 0.4 on line 2. It is concluded that the tap density is related to the content of Co in carbonate precursors NixCoyMn1−x−yCO3. The similar results is also observed on line 3. These results provide a new insight to explore high volumetric energy density cathode materials for advanced LIBs.
| Samples | Tap density (g cm−3) | Samples | Tap density (g cm−3) | Samples | Tap density (g cm−3) |
|---|---|---|---|---|---|
| x = 0,y = 0 | 1.65 | x = 1,y = 0 | 1.59 | x = 0,y = 1 | 1.80 |
| x = 0.2,y = 0 | 1.56 | x = 0.8,y = 0.2 | 1.62 | x = 0,y = 0.8 | 1.77 |
| x = 0.4,y = 0 | 1.62 | x = 0.6,y = 0.4 | 1.66 | x = 0,y = 0.6 | 1.75 |
| x = 0.5,y = 0 | 1.59 | x = 0.5,y = 0.5 | 1.69 | x = 0,y = 0.5 | 1.73 |
| x = 0.6,y = 0 | 1.63 | x = 0.4,y = 0.6 | 1.73 | x = 0,y = 0.4 | 1.72 |
| x = 0.8,y = 0 | 1.61 | x = 0.2,y = 0.8 | 1.77 | x = 0,y = 0.2 | 1.69 |
| x = 1,y = 0 | 1.59 | x = 0,y = 1 | 1.80 | x = 0,y = 0 | 1.65 |
XRD patterns of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) are displayed in Fig. 6. As shown in Fig. 6(a)–(c), all the strong diffraction peaks of oxides could be indexed to a well-defined hexagonal α-NaFeO2-type structure with a space group of Rm. The additional weak reflections at around 2θ = 20°–25° can be attributed to the short-ranged superlattice due to the ordering of Li, Ni, Co, and Mn cations in the transition metal layers.3,4,7–9,17,18 Furthermore, the clear split between the adjacent peaks of (006)/(102) and (018)/(110) suggests a well-developed layered structure for all samples. The lattice parameters of all oxides are calculated, as displayed in Fig. 7. For the samples of line 1, a roughly linear increase in lattice parameters, a and c, can be seen with increase of x value, which may be associated with the change in average ions radius of transitional metals. In line 1, there is substitution equation of 2Ni3+ = Ni2+ + Mn4+. The average ion radius of Ni2+ and Mn4+ is 0.615 Å (the ion radii of Ni2+ and Mn4+ are 0.69 and 0.54 Å, respectively) while the radius of Ni3+ is 0.56 Å.32 Therefore, a larger average ion radius with increasing x value should be responsible for the gradual increase in lattice parameters. On line 2, lattice parameters by contrast linearly decreases with increasing Co content, which can be also explained by the change in average ion radius of transitional metals. As assumed in the introduction, such a substitution equation as Ni2+ + Mn4+ = 2Co3+ exists in line 2. The ion radius of Co3+ (0.53 Å) is smaller than the average one of Ni2+ and Mn4+ (0.615 Å).32 In line 3, a linear increase in lattice parameters with increasing y value is observed, which may be also ascribed to a larger Ni3+ in contrast to Co3+. The linear change in lattice parameter in turn also supports the formation of solid solution among three compounds of Li1.2Ni0.2Mn0.6O2, Li1.2Co0.4Mn0.4O2 and Li1.2Ni0.4Mn0.4O2.
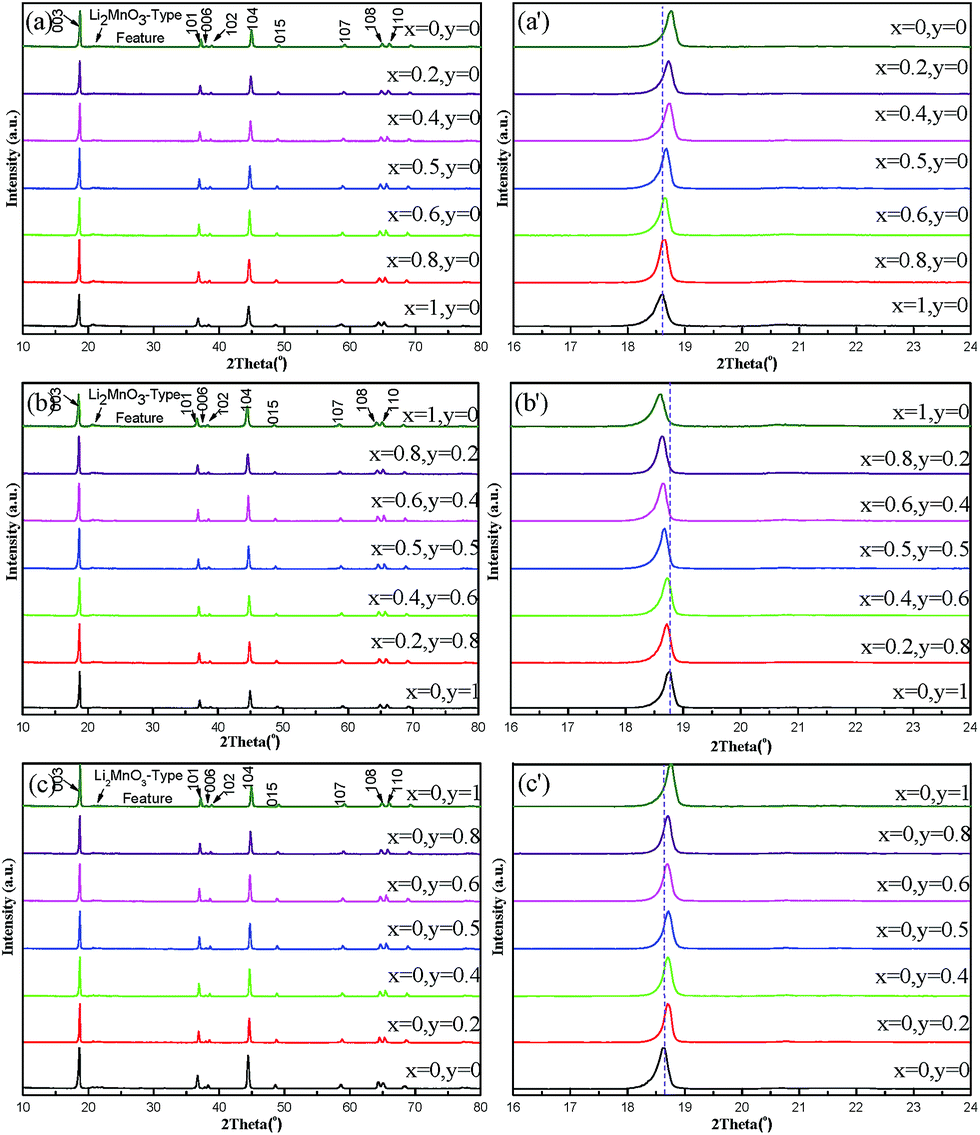 | ||
| Fig. 6 XRD patterns of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1). Full XRD patterns of samples on (a) line 1, (b) line 2 and (c) line 3, respectively. Local magnified XRD patterns near the (003) peak on (a′) line 1, (b′) line 2 and (c′) line 3. | ||
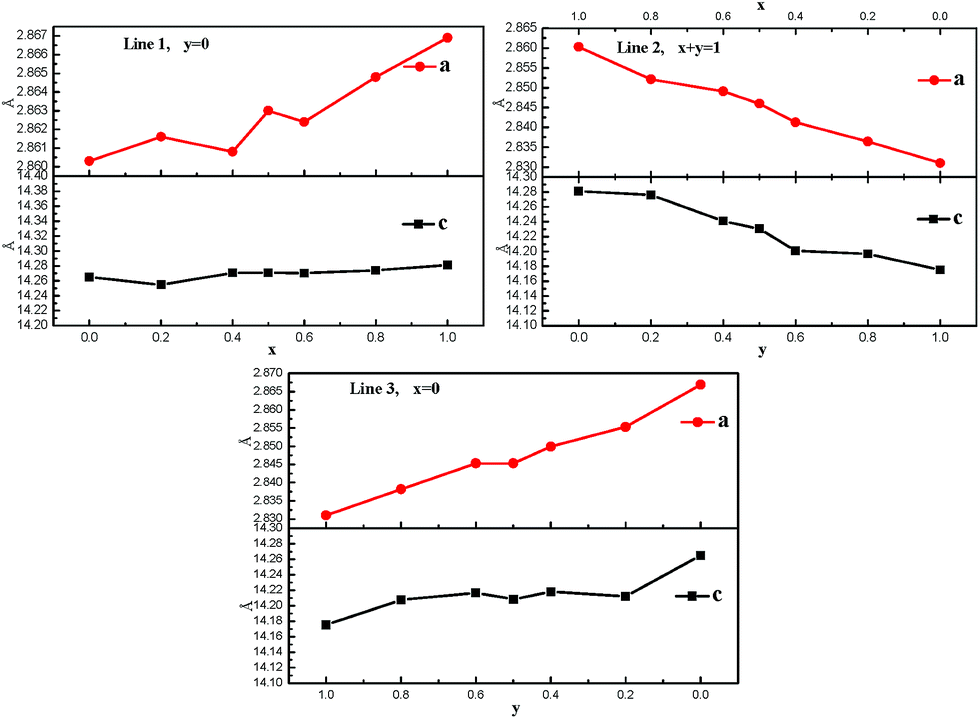 | ||
| Fig. 7 The lattice parameters of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1). | ||
SEM images of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) are showed in Fig. 8. Clearly, spherical or ellipsoidal morphologies of secondary particles are remained for these solid solution materials even after being re-crystallized with lithium sources during high temperature calcination process. Spherical cathode materials have many advantages such as higher packing density and smaller specific surface area, which then enhances volumetric energy density and reduces side reactions between cathode particle and electrolyte. After lithiation, several hundreds nanosized primary particles are revealed in Fig. 8. Meanwhile, the introduction of Co seems to cause porous structure. Usually, the primary particle size and surface morphology play important roles in physical and electrochemical properties such as electrolyte wetting, surface resistance, rate properties and tap density.29–31
 | ||
| Fig. 8 The typical SEM images of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1): samples on line 2. | ||
Volumetric energy density, which is highly related on tap density, is a key factor in Li-ion battery system. The tap densities of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) are listed in Table 2. The samples on line 1 show similar tap density of about 2.0 g cm−3. An increased tap density is achieved while enhancing the Co content, and Li1.2Co0.4Mn0.4O2 delivers the highest tap density of 2.12 g cm−3.
| Samples | Tap density (g cm−3) | Samples | Tap density (g cm−3) | Samples | Tap density (g cm−3) |
|---|---|---|---|---|---|
| x = 0,y = 0 | 2.07 | x = 1,y = 0 | 1.98 | x = 0,y = 1 | 2.12 |
| x = 0.2,y = 0 | 1.95 | x = 0.8,y = 0.2 | 2.00 | x = 0,y = 0.8 | 2.11 |
| x = 0.4,y = 0 | 2.01 | x = 0.6,y = 0.4 | 2.03 | x = 0,y = 0.6 | 2.09 |
| x = 0.5,y = 0 | 1.97 | x = 0.5,y = 0.5 | 2.05 | x = 0,y = 0.5 | 2.09 |
| x = 0.6,y = 0 | 2.01 | x = 0.4,y = 0.6 | 2.07 | x = 0,y = 0.4 | 2.07 |
| x = 0.8,y = 0 | 1.99 | x = 0.2,y = 0.8 | 2.09 | x = 0,y = 0.2 | 2.07 |
| x = 1,y = 0 | 1.98 | x = 0,y = 1 | 2.12 | x = 0,y = 0 | 2.07 |
Fig. 9 shows the initial charge–discharge curves of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) in the voltage range of 2.0–4.8 V at a current density of 20 mA g−1. The initial charge–discharge capacities and Coulombic efficiencies of these samples are summarized in Table 3. The charge capacity below 4.5 V is usually attributed to the oxidation of Ni2+/Ni3+ to Ni4+ and Co3+ to Co4+ while the initial plateau region at around 4.5 V is correlated with the simultaneous expulsion of oxygen and Li+.4,11,13,17 In Fig. 9(a), with the increase of x value the capacity below 4.5 V gradually decreases, but the initial charge plateau gradually prolongs, finally indicating gradually increased initial charge capacity. This implies that the samples with Ni2+ are possibly easier to present the reaction of the simultaneous expulsion of oxygen and Li+ than that with Ni3+. At the same time, the discharge capacity also gradually increase with increasing x. However, a discharge plateau at around 2.7 V is observed and gradually becomes long with increasing x. For Li-rich layered material, origin of the voltage plateau at around 2.7 V for the first discharge process was discussed and verified in a recent report.17 It has been proposed that the oxygen molecule generated in the initial charge process at the high voltage plateau is electrochemically reduced with one electron reduction process below 3.0 V during the subsequent discharge process, forming a superoxide, Li2O2.17 The discharge plateau is also similar to that observed in Li–O2 battery system.33,34 In fact, in some early reports,12,24,28 the discharge plateau at around 2.7 V during initial discharge process could be also seen with careful observation and analysis for some Li-rich materials. In this work, the phenomenon become apparent for some samples, which may be possibly associated with such factors as the samples prepared via a co-precipitation of carbonate as well as characterization of electrochemical properties using coin cell method with small amounts of electrolyte. In addition, another abnormal phenomenon is that the jumping of voltage at the plateau of around 2.7 V is observed for some samples. Repeated experiments and using other commercial electrolyte in our lab still cannot eliminate the phenomenon. It is unclear for us at present. Probably, it is associated with the process of ineffective reduction of oxygen.
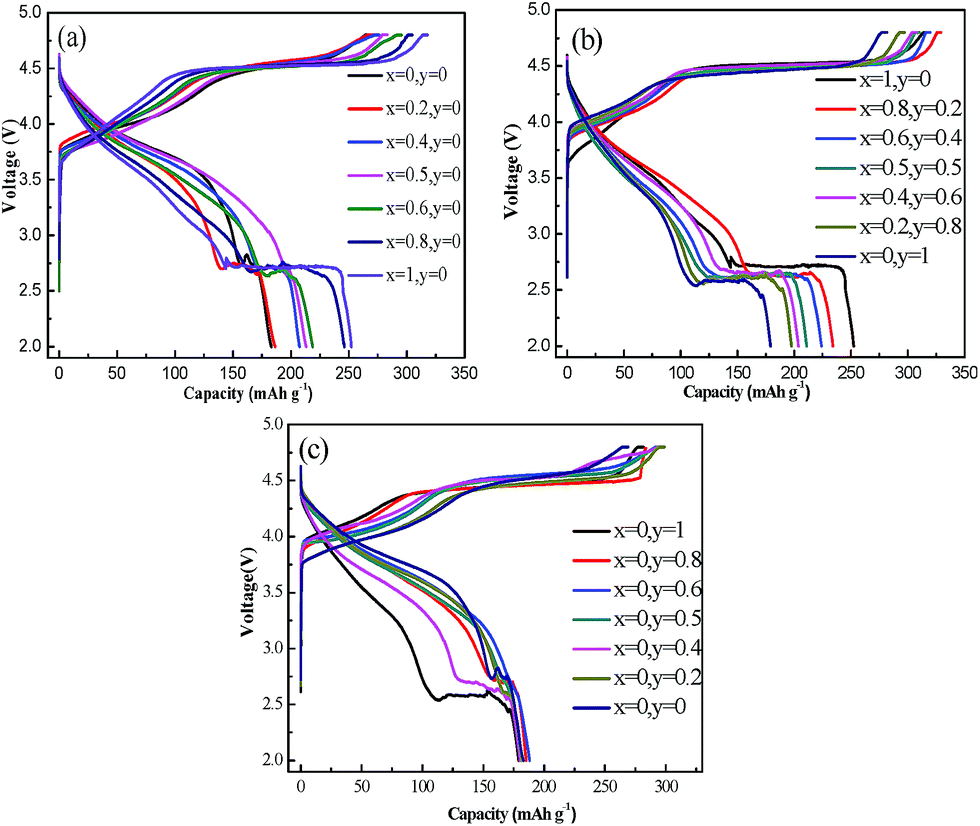 | ||
| Fig. 9 The initial charge–discharge curves of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1): (a) samples of line 1, (b) samples of line 2, (c) samples of line 3. | ||
| Samples | Initial charge capacities (mA h g−1) | Initial discharge capacities (mA h g−1) | Coulombic efficiencies (%) |
|---|---|---|---|
| x = 0,y = 0 | 268.9 | 183.1 | 68.1 |
| x = 0.2,y = 0 | 270.8 | 186.5 | 68.9 |
| x = 0.4,y = 0 | 276.1 | 207.4 | 75.1 |
| x = 0.5,y = 0 | 283.1 | 213.1 | 75.3 |
| x = 0.6,y = 0 | 295.4 | 218.7 | 74.0 |
| x = 0.8,y = 0 | 304.9 | 246.7 | 80.7 |
| x = 1,y = 0 | 318.0 | 252.4 | 79.4 |
| x = 0.8,y = 0.2 | 329.2 | 234.1 | 71.1 |
| x = 0.6,y = 0.4 | 319.6 | 224.1 | 70.1 |
| x = 0.5,y = 0.5 | 310.3 | 211.0 | 68.0 |
| x = 0.4,y = 0.6 | 307.7 | 203.7 | 66.2 |
| x = 0.2,y = 0.8 | 297.1 | 197.6 | 66.5 |
| x = 0,y = 1 | 281.5 | 179.1 | 63.6 |
| x = 0,y = 0.8 | 283.6 | 185.3 | 65.3 |
| x = 0,y = 0.6 | 295.9 | 188.1 | 63.6 |
| x = 0,y = 0.5 | 297.7 | 181.8 | 61.1 |
| x = 0,y = 0.4 | 298.5 | 180.1 | 60.3 |
| x = 0,y = 0.2 | 298.8 | 181.7 | 60.8 |
| x = 0,y = 0 | 268.9 | 183.1 | 68.1 |
As to the materials in line 2, the charge capacities are alike and about 105 mA h g−1 under 4.5 V. Long plateau regions and charge capacities of above 200 mA h g−1 are revealed between 4.5–4.8 V in Fig. 9(b). Nevertheless, the discharge capacity below 3.0 V decreases with the increase of Co3+ content. Compared to these samples in line 1 and 2, the samples in line 3 have shorter plateaus above 4.5 V and lower initial charge–discharge capacities, as shown in Fig. 9(c) and Table 3.
Fig. 10 shows the cycle performances of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) in the voltage rang of 2.0–4.8 V at a current density of 20 mA g−1. The samples on line 1 display superior cycle stability than samples on line 2 and 3. In Fig. 10(a), the sample with x = 0.4 delivers the stable and highest discharge capacity of more than 250 mA h g−1 after initial several cycles. The discharge capacity intends to decrease with a higher Ni content. From Fig. 10(b), it can be concluded that the introduction of Co leads inferior cycling life and lower discharge capacity. Fig. 10(c) also displays that the replacement of Co3+ by Ni3+ can enhance the cycle stability.
 | ||
| Fig. 10 Cycle performances of Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1): (a) samples of line 1, (b) samples of line 2, (c) samples of line 3. | ||
4 Conclusions
A series of layered oxides Li1.2(Ni0.2Mn0.6)x(Co0.4Mn0.4)y(Ni0.4Mn0.4)1−x−yO2 (0 ≤ x + y ≤ 1) have been designed to explore new Li-rich solid solution cathode materials on the basis of tetrahedral phase diagram of LiNiO2–LiCoO2–LiMnO2–Li2MnO3. Micro-sized spherical or ellipsoidal precursors were successfully prepared via a carbonate co-precipitation route. After calcination with lithium sources, the solid solution materials were obtained and all samples can be indexed to a typical layered structure with a Rm space group. The introduction of Co can efficiently improve the tap density of solid solution materials. However, these Co referred samples have lower discharge capacities and suffer from low capacity retention during long cycle. Electrochemical performances closely depend on material compositions. Among these solid solution materials, Li1.2Ni0.3Mn0.5O2 can deliver a high discharge capacity more than 250 mA h g−1 after initial 10 cycles, although a relatively low capacity is observed in initial cycle. More importantly, the activated Li1.2Ni0.3Mn0.5O2 material delivers high discharge capacity of over 230 mA h g−1 even after 70 cycles, indicating superior cycle stability in the range of 2.0–4.8 V. In addition, the apparent reduction of oxygen marked as a discharge plateau at around 2.7 V during the initial discharge process is observed clearly in our work. Thus these results may provide a new insight into explore high-performance cathode materials for advanced LIBs.
Acknowledgements
This work was financially supported by National 863 Program of China (2013AA050906), NSFC (51272175, 21301127).Notes and references
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- S. S. Jan, S. Nurgul, X. Shi, H. Xia and H. Pang, Electrochim. Acta, 2014, 149, 86 CrossRef CAS PubMed.
- Z. Lu, D. D. MacNeil and J. R. Dahn, Electrochem. Solid-State Lett., 2001, 4, A191 CrossRef CAS PubMed.
- Z. Lu and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A815 CrossRef CAS PubMed.
- L. Q. Zhang, K. Takada, N. Ohta, L. Z. Wang, T. Sasaki and M. Watanabe, Mater. Lett., 2004, 58, 3197 CrossRef CAS PubMed.
- J. S. Kim, C. S. Johnson, J. T. Vaughey, M. M. Thackeray, S. A. Hackney, W. Yoon and C. P. Gray, Chem. Mater., 2004, 16, 1996 CrossRef CAS.
- L. Q. Zhang, K. Takada, N. Ohta, K. Fukuda, M. Osada, L. Z. Wang, T. Sasaki and M. Watanabe, J. Electrochem. Soc., 2005, 152, A171 CrossRef CAS PubMed.
- J. Jiang, K. W. Eberman, L. J. Krause and J. R. Dahn, J. Electrochem. Soc., 2005, 152, A1879 CrossRef CAS PubMed.
- J. Jiang and J. R. Dahn, Electrochim. Acta, 2006, 51, 3413 CrossRef CAS PubMed.
- Y. Sun, Y. Xia, Y. Shiosaki and H. Noguchi, Electrochim. Acta, 2006, 51, 5581 CrossRef CAS PubMed.
- A. R. Armstrong, P. Holzapfel, P. Novak, C. S. Johnson, S. H. Kang, M. M. Thackeray and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 8694 CrossRef CAS PubMed.
- J. M. Kim, S. Tsuruta and N. Kumagai, Electrochem. Commun., 2007, 9, 103 CrossRef CAS PubMed.
- T. A. Arunkumar, Y. Wu and A. Manthiram, Chem. Mater., 2007, 19, 3067 CrossRef CAS.
- C. S. Johnson, N. Li, C. Lefief, J. T. Vaughey and M. M. Thackeray, Chem. Mater., 2008, 20, 6095 CrossRef CAS.
- J. M. Zheng, Z. R. Zhang, X. B. Xu, Z. X. Dong, Z. Zhu and Y. Yang, J. Electrochem. Soc., 2008, 155, A775 CrossRef CAS PubMed.
- I. Atsushi, D. C. Li and Y. Sato, J. Power Sources, 2010, 195, 567 CrossRef PubMed.
- N. Yabuuchi, K. Yoshii, S. T. Myung, I. Nakai and S. Komaba, J. Am. Chem. Soc., 2011, 133, 4404 CrossRef CAS PubMed.
- K. A. Jarvis, Z. Q. Deng, L. F. Allard, A. Manthiram and P. J. Ferreira, Chem. Mater., 2011, 23, 3614 CrossRef CAS.
- J. Bareno, M. Balasubramanian, S. H. Kang, J. G. Wen, C. H. Lei, V. Pol, I. Petrov and D. P. Abraham, Chem. Mater., 2011, 23, 2039 CrossRef CAS.
- L. Li, Y. L. Chang, H. Xia, B. H. Song, J. R. Yang, K. S. Lee and L. Lua, Solid State Ionics, 2014, 264, 36 CrossRef CAS PubMed.
- X. Huang, M. Wang and R. Che, J. Mater. Chem., 2014, 2, 9656 RSC.
- J. Ma, Y. N. Zhou, Y. Gao, Q. Kong, Z. Wang, X. Q. Yang and L. Chen, Chem. – Eur. J., 2014, 20, 8723 CrossRef CAS PubMed.
- J. Lu, Y. Chang, B. Song, H. Xia, J. Yang, K. S. Lee and L. Lu, J. Power Sources, 2014, 271, 604 CrossRef CAS PubMed.
- L. Q. Zhang, Q. W. Peng, Z. W. Lu, L. B. Ren, H. Q. Ren, X. J. Liu, The 15th International Meeting on Lithum Batteries, Abstract #629, 2010, June 27-July 3, Montreal, Canada.
- L. Q. Zhang, C. W. Xiao and R. J. Yang, Prog. Chem., 2011, 23, 410 CAS.
- P. S. Whitfield, S. Niketic and I. D. Davidson, J. Power Sources, 2005, 146, 617 CrossRef CAS PubMed.
- L. Q. Zhang, X. Q. Wang, T. Muta, D. C. Li, H. Noguchi, M. Yoshio, R. Z. Ma, K. Takada and T. Sasaki, J. Power Sources, 2006, 162, 629 CrossRef CAS PubMed.
- L. Q. Zhang, H. Noguchi, D. C. Li, T. Muta, X. Q. Wang, M. Yoshio and I. Taniguchi, J. Power Sources, 2008, 185, 534 CrossRef CAS PubMed.
- D. K. Lee, S. H. Park and K. Amine, J. Power Sources, 2006, 162, 1346 CrossRef CAS PubMed.
- H. Deng, I. Belharouak, R. E. Cook, H. Wu, Y. K. Sun and K. Amine, J. Electrochem. Soc., 2010, 157, A447 CrossRef CAS PubMed.
- D. P. Wang, I. Belharouak, G. M. Koenig, G. Zhou and K. Amine, J. Mater. Chem., 2011, 21, 9290 RSC.
- R. D. Shannon, Acta Crystallogr., 1976, A32, 751 CrossRef CAS.
- Z. Peng, S. A. Freunberger, Y. Chen and P. G. Bruce, Science, 2012, 337, 563 CrossRef CAS PubMed.
- Z. Peng, S. A. Freunberger, L. J. Hardwick, Y. Chen, V. Giordani, F. Bardé, P. Novák, D. Graham, J. M. Tarascon and P. G. Bruce, Angew. Chem., Int. Ed., 2011, 50, 6351 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |