
Environmentally benign synthesis of vinyl ester resin from biowaste glycerin†
Priyank N. Shah*a,
Namjoon Kimc,
Zhuangrong Huangc,
Mahesh Jayamannaa,
Akshay Kokila,
Alex Pineb,
Jarmin Kaltsasb,
Edwin Jahngena,
David K. Ryana,
Seongkyu Yoonc,
Robert F. Kovard and
Yongwoo Lee*a
aDepartment of Chemistry, University of Massachusetts Lowell, One University Avenue, Lowell, Massachusetts 01854, USA. E-mail: Priyank_shah@uml.edu; Yongwoo_Lee@uml.edu; Tel: +1-978-934-3734 Tel: +1-978-934-3792
bMaine Standard Biofuels, 51 Ingersoll Dr., Portland, Maine 04103, USA
cDepartment of Chemical Engineering, University of Massachusetts Lowell, One University Avenue, Lowell, Massachusetts 01854, USA
dR. F. Kovar & Associates, 203 Beach Street, Wrentham, MA 02093-1513, USA
First published on 22nd April 2015
Abstract
We present here for the first time a novel environmentally benign protocol for the synthesis of vinyl ester resin (VER). Our synthetic strategy utilizes a commercial waste material, glycerin, from biodiesel manufacturing and converts it into a widely utilized resin. The VER was synthesized using bisphenol A (BPA) and glycidyl methacrylate (GMA) as precursors. GMA was synthesized via a multistep synthetic protocol using glycerin obtained from a biodiesel manufacturing waste stream. The structure of the intermediates was confirmed by 1H NMR, HPLC and FT-IR spectroscopy.
Introduction
Vinyl ester resin (VER), introduced in the late 1960s, is a low-cost material that can be processed to form a matrix of fiber-reinforced composites that are excellent materials for hulls, transportation vehicles, and many other structural composites.1 Fiber-reinforced composites produced from VER have in particular become the materials of choice for marine applications. VER is therefore finding increasing applications in transportation, construction, marine, and wind energy industries.2–5 According to recent investigations, BPA has been shown as an endocrine disrupter, has been found to bind to estrogen receptors and have estrogenic effects.6 Humans are exposed to BPA majorly through their diet as the food might be in contact with containers or food packaging made with un-crosslinked polymers containing BPA as the repeat units. Vinyl ester resin typically contains BPA, because the chemical structure has displayed to yield the best mechanical performance of the composites used mostly in the marine industry for fabrication of hulls. We chose to utilize BPA as a building block for the environmentally benign synthesis of VER resin to display the feasibility of this approach, which will be extended to other non-BPA building blocks.VER can be processed at ambient temperature using vacuum-assisted resin transfer molding (VARTM) into massive carbon-fiber-reinforced composites.5 The synthetic protocols used for manufacture of VER has two main disadvantages. First, VER is currently synthesized from petroleum feedstock, which reduces the sustainability. Second, VER is synthesized from epichlorohydrin which is a known environmental hazard that produces acute toxicity in case of inhalation, oral, and dermal exposure.
Hence, for reducing the environmental impact, it is essential to develop VER synthesis routes that are not dependent on petroleum based feedstock. Moreover, investigations of the VER synthesis routes that lead to elimination of the use of hazardous chemicals, such as epichlorohydrin, are necessary. To assure sustainability of the new processing route, starting materials should be derived from readily available renewable resources.
Recently, significant advances have been made for obtaining biodiesel, a renewable biofuel for use in diesel engines and heating applications. Biodiesel can be produced by base catalyzed hydrolysis of rapeseed oil,7 soybean and wastes of cooked oil,8 rice straw,9 vegetable oil mixtures of cottonseed, soybean and castor oils,10 jatropha oil,11 and crude palm oil.12 Biodiesel is environmentally beneficial because it is biodegradable, nontoxic and has low emission.13 During manufacturing of biodiesel, approximately a 10% volume of glycerin is produced as a by-product or waste.14 Therefore, the effective utilization of glycerin enlarges the economy of biodiesel production; moreover, the conversion of glycerin to value added chemicals can expand the scope of green chemistry.15 Although biodiesel uses non-petroleum feedstocks, it is still subject to the price fluctuations of the petroleum liquid fuels market. Subsidization also plays an important role in the overall market price of biodiesel. These two market factors often make it very important for biodiesel manufacturers to sell as many of their byproducts as possible.
Only limited systematic investigations are reported on the use of functional vinyl monomer such as glycidyl methacrylate (GMA),16 which is a versatile bidentate monomer capable of imparting oxirane functionality to VER. It is also worth mentioning that bisphenol A (BPA) is an important chemical used to manufacture polycarbonate plastic, epoxy resin, flame retardants, and other specialty products,17 and is manufactured by acid catalyzed condensation of acetone and phenol.18
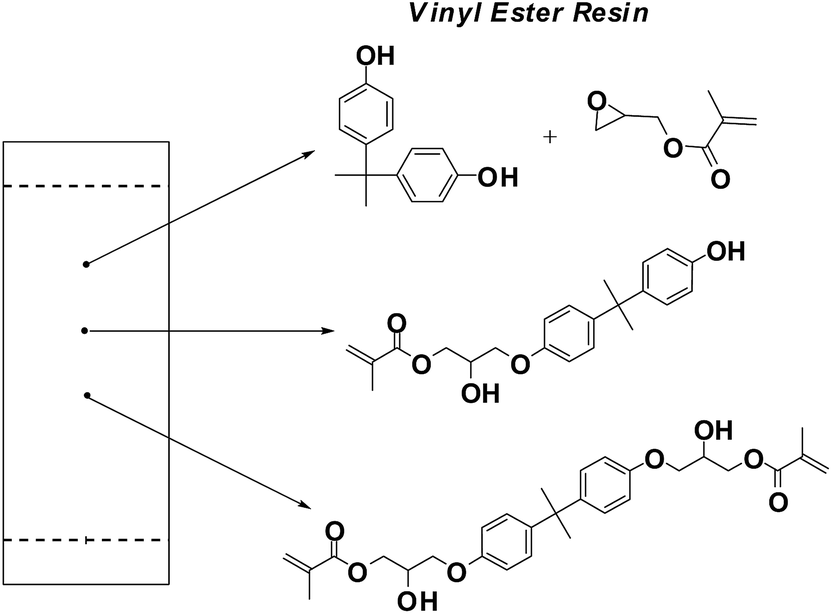
In the present work, we focused on the synthesis of the green vinyl ester resins (GVER) from renewable sources using three steps (Fig. 1): (1) synthesis of GMA from waste glycerin; (2) conversion of phenol into BPA using environmentally benign conditions, and (3) synthesis of GVER from BPA and GMA.
 | ||
| Fig. 1 Synthesis of vinyl ester oligomer from phenol and glycerin. | ||
Experimental section
Materials
Crude glycerin (Maine Standard Biofuels), potassium carbonate (99% Sigma-Aldrich), dimethyl carbonate (99% Alfa-Aesar), sodium sulfate (99.9% Alfa-Aesar), methyl methacrylate (99.9% Alfa-Aesar), potassium cyanide (Sigma-Aldrich), 2,4-dimethyl-6-tert-butyl phenol (Alfa-Aesar), phenol (99% Sigma-Aldrich), acetone (99.9% Sigma-Aldrich), Amberlyst® 15 hydrogen form (Sigma-Aldrich), 2-diethylaminoethanethiol hydrochloride (95% Sigma-Aldrich) and N,N-dimethylbenzylamine were used as received.Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 mixture of hexane and ethyl acetate. Samples were prepared in ethyl acetate.
:30 mixture of hexane and ethyl acetate. Samples were prepared in ethyl acetate.Glycerol carbonate. In a typical setup, a 250 mL flask equipped with a magnetic stirrer, condenser and thermometer was charged with glycerin (40.05 g, 0.435 mol), dimethyl carbonate (117.45 g, 1.305 mol) and K2CO3 (1.8 g, 13.05 mmol). The reaction mixture was refluxed (73–75 °C) for 3 h. After completion of the reaction, methanol and the excess of dimethyl carbonate were distilled off at 40 °C under reduced pressure. The remaining glycerol carbonate was analyzed by 1H NMR and FT-IR spectroscopy. Glycerol carbonate was isolated with 98.5% conversion. Glycerol carbonate: 1H NMR (DMSO, 500 MHz), δ (ppm), 5.20 (1H, –OH), 4.80 (1H, –CH2–CH–), 4.49, 4.31 (2H, –O–CH2), 3.67, 3.53 (2H, –CH2–CH–). 13C NMR (DMSO, 125 MHz) δ (ppm): 155.97 (CO–O–), 77.17 (–OH–CH2–CH–), 65.87 (–OH–CH2–CH–), 60.80 (–OH–CH2–CH–CH2). IR (cm−1): 3428 (–OH stretching), 2920 (–CH stretching aliphatic), 1761 (–C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1480, 1399, 1277 (–CH bending).
O stretching), 1480, 1399, 1277 (–CH bending).
Glycidol. A distillation apparatus with a 100 mL flask was placed in an oil bath with magnetic stirring. The flask was charged with 40.0 g of the glycerol carbonate and 4.0 g of anhydrous sodium sulphate. The reaction mixture was continuously stirred using magnetic stirrer under reduced pressure and the flask was heated to 160 °C. Glycidol was allowed to be distilled off for 3 h. The success of the reactions (i.e., purity of both glycerol carbonate and glycidol) were determined by H1 NMR and FTIR. Glycidol was isolated in pure form with 33.0% yield. Glycidol: 1H NMR (CDCl3, 500 MHz), δ (ppm), 4.83 (1H, –OH), 2.99 (1H, –CH2–CH–), 3.60, 3.33 (2H, –O–CH2), 2.68, 2.53 (2H, –CH2–CH–). 13C NMR (CDCl3, 125 MHz) δ (ppm): 62.13 (–OH–CH2–CH–), 52.44 (–OH–CH2–CH–), 44.30 (–OH–CH2–CH–CH2). IR (cm−1): 3420 (–OH stretching), 2926 (–CH stretching aliphatic), 1397, 1269 (–CH bending).
Glycidyl methacrylate. A flask was charged with 50.0 g (0.5 moles) of methyl methacrylate; 7.4 g (0.1 mol) of glycidol; 0.05 grams of 2,4-dimethyl-6-tert butyl phenol as polymerization inhibitor; and 0.055 g (0.85 millimoles) of potassium cyanide and the mixture heated to 70–80 °C for 2 h. They were immediately vacuum distilled and two fractions were collected. Glycidyl methacrylate was isolated with 25.3% yield. Glycidyl methacrylate: 1H NMR (CDCl3, 500 MHz), δ (ppm), 6.09, 5.72 (2H, –C
CH2), 4.48, 3.92 (2H, –CH2–CH–), 3.25 (1H, –CH2–CH–), 2.82, 2.67 (2H, –O–CH2), 1.90 (3H, –C–CH3). 13C NMR (CDCl3, 125 MHz) δ (ppm): 166.35 (C–CO–O–), 135.70 (–CH2C–CO–O), 125.51 (–CH2C–CO–O), 64.92 (–O–CH2–epoxy ring), 48.93 (–O–CH2–CH (epoxy ring)), 43.96 (–O–CH2 (epoxy ring)), 17.78 (–CH2C–CH3). IR (cm−1): 2995, 2951 (–CH stretching aliphatic), 1726 (–CO stretching), 1452, 1397, 1326, 1269 (–CH bending).
Synthesis of bisphenol A from phenol
A 50 mL flask was charged with phenol (10.0 g, 106.3 mmol), 100 mg amberlyst-15, 187.75 mg of 2-diethylamino-ethanethiol hydrochloride and acetone (2.80 g, 48.31 mmol). The reaction mixture was heated to 65–90 °C for 24 h with constant stirring. After 24 hours, the reaction mixture was cooled to ambient temperature, and the product was analysed by TLC and 1H NMR. The BPA was purified by column chromatography using 3:7 ethyl acetate:hexane ratio. The 1H NMR showed that the conversion of BPA from phenol was ∼27.0%, but after column purification, we could successfully isolated BPA with 9.0% yield. Bisphenol A: 1H NMR (DMSO, 500 MHz), δ (ppm), 9.12 (2H, –OH), 6.98, 6.62 (8H, –ArH), 1.54 (6H, –C–CH3). 13C NMR (DMSO, 125 MHz) δ (ppm): 155.39 (OH–C(ArC)), 141.57 (–CH3–C–C(ArC)), 127.7 (–CH3–C–C(ArC)–C(ArC)), 155.08 (OH–C(ArC)–C(ArC)), 41.38 (–CH3–C–C(ArC)), 31.40 (–CH3–C–C(ArC)). IR (cm−1): 3320 (–OH stretching), 2963 (–CH stretching Ar), 1508, 1434, 1383 (–CH bending).
Synthesis of vinyl ester resins (VER)
A 25 mL RB flask was charged with BPA (1.14 g, 5 mmol), GMA (1.42 g, 10 mmol) and N,N-dimethylbenzylamine (0.0065 g, 0.05 mmol). The flask had continuous flow of argon, and the temperature was raised to 70 °C, which was maintained for 6 h. After the reaction, the highly viscous liquid was transferred to a glass vial, and the product was analysed by TLC, 1H NMR, and FT-IR. By HPLC, the conversion of difunctional VER was observed to be 69.3%. Difunctional bisphenol A: 1H NMR (CDCl3, 500 MHz), δ (ppm), 6.09, 5.72 (2H, –CCH2), 4.48, 3.92 (2H, –CH2–CH–), 3.25 (1H, –CH2–CH–), 2.82, 2.67 (2H, –O–CH2), 1.90 (3H, –C–CH3). 13C NMR (DMSO, 125 MHz) δ (ppm): 167.09 (C–CO–O–), 156.58 (–CH2–O–C(ArC)), 143.25 (–CH3–C–C(ArC)), 136.13 (–CH2C–CO–O), 127.78 (–CH2C–CO–O), 126.45 (–CH2–O–C(ArC)–C(ArC)), 114.10 (–CH3–C–C(ArC)–C(ArC)), 69.27 (CO–O–CH2–CH– CH2), 67.35 (CO–O–CH2–CH–CH2), 66.01 (CO–O–CH2–CH–CH2), 39.48 (–CH3–C–C(ArC)), 30.78 (–CH3–C–C(ArC)), 18.35 (–CH2C–CH3). IR (cm−1): 3424 (–OH stretching), 2963, 2926 (–CH stretching aliphatic), 1724 (–CO stretching), 1634, 1607, 1597, 1403, 1362 (–CH bending).
Results and discussion
Here we demonstrate the sustainable, environmentally benign synthesis of ‘green’ vinyl ester resin (GVER). The GVER was synthesized predominantly from a bio-waste, namely glycerin, recovered from biodiesel waste stream. Such source materials are renewable, low-cost, and are becoming increasingly abundant and thus are expected to significantly reduce the dependence on petroleum feedstock.Synthesis of glycidyl methacrylate from glycerin
GMA was synthesized using glycerin as the main precursor. Glycerin is a side-product of the transesterification of triglyceraldehyde in manufacturing of biodiesel or the hydrolysis of triglyceraldehyde during saponification. Fig. 2 demonstrates the mixture of products generated during the synthesis of biodiesel at Maine Standard Biofuels (MSB), which has biodiesel in the top layer, soaps in the middle, and waste glycerin in the bottom. We obtained the glycerin from waste stream of a biodiesel production facility of MSB. The crude glycerin was purified using vacuum distillation at 190 °C, and isolated in pure form as shown in Fig. 3. | ||
| Fig. 2 Small scale biodiesel reaction at Maine Standard Biofuels (MSB). Biodiesel/methyl esters (top), soaps (middle), crude glycerin (bottom). | ||
 | ||
| Fig. 3 1H NMR of the distilled glycerin. | ||
Glycerol carbonate (GC), a first intermediate in the synthetic route to GMA from the distilled glycerin, is an attractive intermediate having a large number of applications in various fields. Various approaches had been adopted by different groups for the synthesis of the GC; such as, using (a) Novozyme 435 catalyst and dimethyl carbonate as a precursor,17 and (b) metal oxide as a catalyst and urea as a precursor.18 In a new approach, GC was synthesized by reaction between glycerin and dimethyl carbonate at 73 °C for 3 h in the presence of potassium carbonate.19 The method is promising, with several challenges for example the separation of potassium carbonate from the reaction mixture. Firstly, the reaction was performed at 73 °C and the GC was synthesized successfully. The product formation was confirmed by 1H NMR as shown in Fig. 4. The peak integration value suggested ∼98.5% conversion of glycerin to GC at 73 °C. In order to complete the reaction to 100.0%, the reaction temperature was increased to 100 °C; however, as the temperature increased to 100 °C, formation of some side product was observed and unreacted glycerin was also found in the final mixture. The FT-IR spectra of glycerol carbonate synthesized at 73 °C clearly showed peaks of –CO at 1772.51 cm−1 and –OH at 3456.9 cm−1 as shown in Fig. S2 in ESI.†
 | ||
| Fig. 4 1H NMR of glycerol carbonate and glycidol. | ||
Glycidol is the second intermediate during the synthesis of GMA from glycerin. Glycidol was obtained by decarboxylation of GC as shown in Fig. 1. In a typical setup for the synthesis of glycidol, a vacuum distillation apparatus was utilized. Glycidol was synthesized by heating the glycerol carbonate in the presence of sodium sulfate at 180 °C for 3 h.20 The formation of glycidol was confirmed by 1H NMR by matching the integration value of each proton as shown in Fig. 4. Strong evidence is provided by the FT-IR exhibiting a clear disappearance of the –CO peak at 1761 cm−1 as shown in Fig. S2 in ESI.† Glycidol was isolated with 33.0% yield.
Glycidyl methacrylate (GMA) is one of the precursors for the synthesis of vinyl ester resin monomer along with bisphenol A. GMA was synthesized by transesterfication between methyl methacrylate and the oxirane moiety in the presence of potassium cyanide as a catalyst and 2,4-dimethyl-6-tertbutylphenol.21 First, the pressure was reduced in the reaction flask, and the temperature was raised to 70 °C. The temperature of 70 °C was maintained for 2 h, and after that unreacted methyl methacrylate and methanol were distilled off. In a second fraction unreacted glycidol was collected and in last fraction GMA was obtained. The structure of GMA was confirmed by 1H NMR as shown in Fig. 5. The GMA was isolated with 25.3% yield.
 | ||
| Fig. 5 1H NMR of glycidyl methacrylate. | ||
Synthesis of bisphenol A (BPA)
BPA was synthesized using a solvent-free environmentally benign synthetic protocol, in which phenol was first melted in an oil bath at 60 °C then combined with acetone in the presence of Amberlyst 15 as the catalyst and 2-diethylaminoethanethiol hydrochloride as a promoter.16 The reaction was performed in a reactor sealed with a plastic stopper. The mixture yielded a crude product subsequently identified as a mixture of BPA and unreacted phenol using thin layer chromatography and 1H NMR. The synthesized BPA was purified by column chromatography using hexane:ethyl acetate in a 7:3 ratio as a solvent mixture. The BPA was isolated with 9.0% yield and product was confirmed by 1H NMR as shown in Fig. 6.
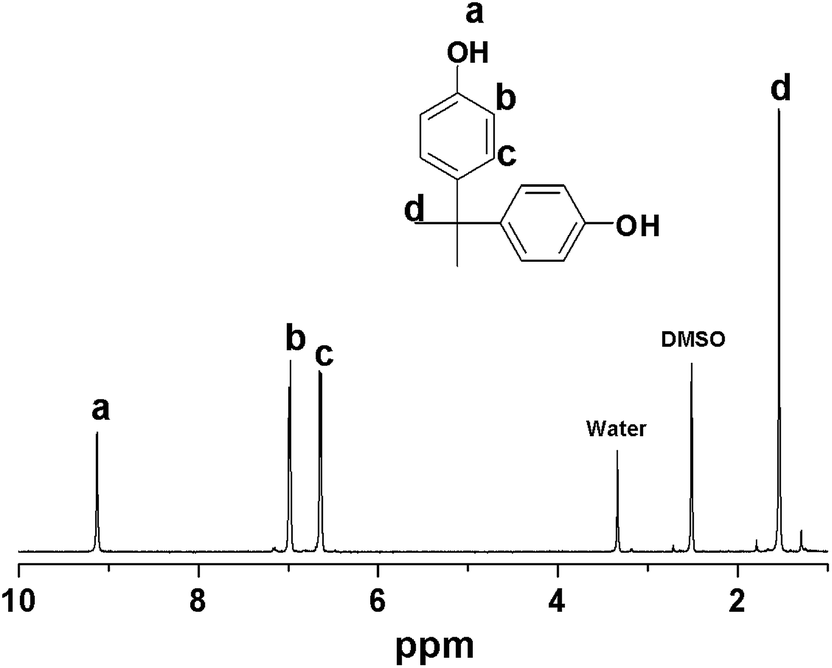 | ||
| Fig. 6 1H NMR of bisphenol A. | ||
Synthesis of vinyl ester resin (VER) monomer
VER are formulated from VER monomer using a solvent/reactive diluent, typically styrene, as well as additives to meet specific needs. In general, VER has several advantages over other polymers. For example, toughened and brominated VER produces resins with higher strengths than polyesters and lower viscosity than epoxies. This low viscosity is critical for fabricating large and intricate structures by vacuum infusion. Fiberglass-reinforced VER composites are preferred for many structural composite applications. Recent advances in compatible sizing have enabled stiffer reinforcement of VERs using carbon fiber.The VER monomer was synthesized by a reaction between BPA and GMA in the presence of amine catalyst. The reaction was performed at 70 °C under argon.22 The 1H NMR did not exhibit the –OH peak of the BPA, so it was presumed that the reaction was 100% complete but, the integration value did not matching precisely. Thin layer chromatography (TLC) displayed three spots. The integration values in the 1H NMR and the higher number of spots in TLC confirmed the presence of product in the reaction mixture as shown in Fig. 7.
 | ||
| Fig. 7 TLC of vinyl ester resin mixture in ethyl acetate and hexane in 3:7 ratios. | ||
After confirming the presence of three different products in the reaction mixture; all fraction were separated by column chromatography using a 3:7 ethyl acetate:hexane ratio and analysed using 1H NMR as shown in Fig. 8. The 1H NMR clearly indicated the presence of non-functional BPA (NF) in the first fraction; mono-functional BPA (MF), GMA-BPA, in the second fraction; and di-functional BPA (DF), GMA-BPA-GMA, in the third fraction. The data clearly suggested that a 2 h reaction time was insufficient to obtain the maximum amount of DF. In order to increase the yield the reaction time was increased up to 6 h and sample was taken out at periodic interval of time, and the molar ratio of the GMA:BPA was increased to 4:1. A very strong evidence of the formation of the vinyl ester resin can be observed in the shifting of the aromatic peaks in 1H NMR of the vinyl ester resin as shown in Fig. S9 in the ESI.† The peaks for the BPA aromatic protons can be observed at 7.10 (OH–C–CH(ArH)) and 6.83 (CH–C–C–CH3), which shift to 6.98 (–O–C–CH) and 6.63 (CH–C–C–CH3), respectively. The peak for all four aromatic protons can be observed at 7.10 (OH–C–CH(ArH)), 6.98 (–O–C–CH), 6.83 (CH–C–C–CH3), and 6.63 (CH–C–C–CH3) as shown in Fig. S9 in the ESI.†
 | ||
| Fig. 8 1H NMR of each fraction from column separation. | ||
In order to determine the kinetics of the reaction, the quantification of NF, MF and DF in the reaction mixture is very essential. Such quantification can be obtained by high performance liquid chromatographic (HPLC) separation of the mixture. Therefore, preliminary work was conducted to develop a HPLC method for the separation and determination of BPA and reaction products in the synthesis of a VER. The ultimate goal was to separate each fraction without overlapping retention time.
Chromatograms shown in Fig. S4–S6 in ESI† clearly demonstrate that the 3 compounds have very different retention times on the diol column with 7:3 hexane:ethyl acetate and should be easily separated and determined in a reaction mixture. The first fraction (NF), second fraction (MF) and third fraction (DF) exhibited retention time of 5.218, 7.946, and 13.686 min respectively.
We quantified the NF, MF, and DF BPA by calculating and comparing the area under the peak of the chromatogram obtained at different time intervals as shown in Table 1. The results suggested that reaction proceeds well as time progresses. However, it did not attain completion at 6 h, 69.26% of the DF VER monomer was obtained. Experiments are in progress and the results will be published in near future with additional work to further improve the yield of this synthesis.
| Reaction time (h) | Different functional BPA | ||
|---|---|---|---|
| Non | Mono | Di | |
| 2 h | 32.00 | 49.24 | 18.76 |
| 3 h | 20.79 | 46.28 | 32.93 |
| 4 h | 14.13 | 40.63 | 45.23 |
| 5 h | 13.38 | 28.27 | 58.34 |
| 6 h | 12.70 | 18.05 | 69.26 |
Conclusions
Glycidyl methacrylate, a valuable monomer in its own right, was successfully synthesized using glycerin from biodiesel waste stream and environmentally benign reaction conditions. During the novel synthetic protocol other valuable intermediates, such as glycerol carbonate and glycidol, were also obtained. The second precursor, bisphenol A was synthesised by the reaction of phenol and acetone using an environmentally benign synthetic protocol. The GVER was synthesized by reaction of BPA and GMA. This synthetic strategy opens avenues for obtaining this valuable resin using sustainable environmentally benign routes.Acknowledgements
The author would like to thank the Strategic Environmental Research and Development Program (SERDP WP-1755; WP-2317) for financial support of this work.References
- J. S. Martin, J. M. Laza, M. L. Morrás, M. L. Rodríguez and M. León, Polymer, 2000, 41, 4203 CrossRef CAS.
- A. L. Nazareth da Silva, S. C. S. Teixeira, A. C. C. Widal and F. M. B. Coutinho, Polym. Test., 2000, 20, 895 CrossRef.
- D. Rosu, L. Rosu and C. N. Cascaval, J. Photochem. Photobiol., A, 2008, 194, 275 CrossRef CAS PubMed.
- M. Sultania, S. B. Yadaw, J. S. P. Rai and D. Srivastava, Mater. Sci. Eng., A, 2010, 527, 4560 CrossRef PubMed.
- (a) C. F. Leitten Jr, W. L. Griffith, A. L. Compere and J. T. Shaffer, Society of Automotive Engineers, 2010, 02FCC-144 Search PubMed; (b) W. L. Griffith, C. F. Leitten Jr, J. T. Shaffer and A. L. Compere, Low Cost Carbon Fiber from Renewable Resources, Oak Ridge National Laboratory Report, 1999 Search PubMed; (c) E. Sarlin, M. Lindgren, R. Suihkonen, S. Siljander, M. Kakkonen and J. Vuorinen, Wear, 2015 DOI:10.1016/j.wear.2015.03.021.
- J. R. Rochester, Reprod. Toxicol., 2013, 42, 132 CrossRef CAS PubMed.
- M. Hájek, F. Skopal, L. Čapek, M. Černoch and P. Kutálek, Energy, 2012, 48, 392 CrossRef PubMed.
- M. Koberg, R. Abu-Much and A. Gedanken, Bioresour. Technol., 2011, 102, 1073 CrossRef CAS PubMed.
- L. Zheng, Y. Hou, L. Wu, S. Yang, Q. Li and Z. Yu, Energy, 2012, 47, 225 CrossRef CAS PubMed.
- S. M. P. Meneghetti, M. R. Meneghetti, T. M. Serra, D. C. Barbosa and C. R. Wolf, Energy Fuels, 2007, 21, 3746 CrossRef CAS.
- H. V. Lee, Y. H. Taufiq-Yap, M. Z. Hussein and R. Yunus, Energy, 2013, 49, 12 CrossRef CAS PubMed.
- Z. Man, Y. A. Elsheikh, M. A. Bustam, S. Yusup, M. I. Abdul Mutalib and N. Muhammad, Ind. Crops Prod., 2013, 41, 144 CrossRef CAS PubMed.
- G. Chen and B. Fang, Bioresour. Technol., 2011, 102, 2635 CrossRef CAS PubMed.
- M. R. Nanda, Z. Yuan, W. Qin, H. S. Ghaziaskar, M. Poirier and C. C. Xu, Fuel, 2014, 117, 470 CrossRef CAS PubMed.
- P. S. Reddy, P. Sudarsanam, B. Mallesham, G. Raju and B. M. Reddy, J. Ind. Eng. Chem., 2011, 17, 377 CrossRef CAS PubMed.
- (a) R. A. Reinicker and B. C. Gates, AIChE J., 1974, 20, 933 CrossRef CAS PubMed; (b) L. Hou, Q. Cai, B. Lu, X. Li, X. Xiao, Y. Han and S. Cui, Catal. Lett., 2006, 111, 153 CrossRef CAS PubMed.
- S. C. Kim, Y. H. Kim, H. Lee, D. Y. Yoon and B. K. Song, J. Mol. Catal. B: Enzym., 2007, 49, 75–78 CrossRef CAS PubMed.
- L. Wang, Y. Ma, Y. Wang, S. Liu and Y. Deng, Catal. Commun., 2011, 12, 1458 CrossRef CAS PubMed.
- G. Rokicki, P. Rakoczy, P. Parzuchowski and M. Sobiecki, Green Chem., 2005, 7, 529 RSC.
- V. A. Currier, J. B. Bell Jr and J. D. Malkemus, Method for preparing glycidol, US pat., 2,856,413, Oct. 14, 1958.
- R. Ackerman, H. Kolb, H. Morlock and G. Schreyer, Process for the production of glycidyl methacrylate, US pat., 4,228,084, Oct. 14, 1980.
- R. L. Bowen, Method of preparing a monomer having phenoxy and methacrylate groups linked by hydroxy glyceryl groups, US pat., 3179623, Apr. 20, 1965.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03254g |
| This journal is © The Royal Society of Chemistry 2015 |