
Physical and chemical studies of tungsten carbide catalysts: effects of Ni promotion and sulphonated carbon†
Abstract
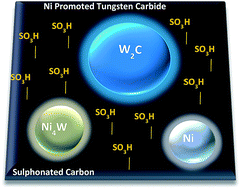
Ni promoted tungsten carbides have been shown to be an effective catalyst for cellulose conversion reaction. With the use of both in situ and ex situ techniques an investigation into the physical and chemical aspects of the Ni-promoted tungsten carbide catalyst supported on activated carbon either in pure form or functionalized with sulfuric acid was conducted. In situ XRD analysis performed during the carburization process showed that non-promoted samples formed a mixture of nanosized W2C, WC1−x and WC carbide phases. In the case of Ni promoted catalysts, in situ XRD, XANES, XPS and TEM analysis revealed that Ni aids in lowering the carburization temperature by 50 °C but also assisted in the deposition of polymeric carbon onto the catalyst surface which reduced cellulose conversion. However, the results indicate beneficial effects caused by the high carbon coverage by stopping the W2C to WC carbide phase transition. Thus, carburization of Ni promoted samples produced only W2C phase, which is stable up to 800 °C. The functionalization of activated-carbon with –SO3H not only increases the hydrolysis of cellulose but also lead to a greater dispersion of Ni over the catalyst. The resulting improvement in the interaction between Ni/W/C increases the cellulose transformation in a one-pot synthesis towards the production of ethylene glycol.
 Please wait while we load your content...
Please wait while we load your content...

