
Rapid microwave assisted hydrothermal synthesis of porous α-Fe2O3 nanostructures as stable and high capacity negative electrode for lithium and sodium ion batteries
Abstract
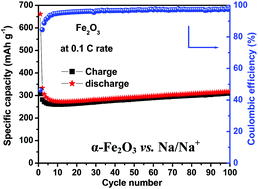
Hematite porous α-Fe2O3 nanostructures were prepared within 60 minutes by a rapid microwave assisted hydrothermal process. X-ray diffraction (XRD) and Fourier transform infrared (FTIR) spectroscopy studies confirm the phase and structural co-ordination of α-Fe2O3, respectively. Scanning electron microscopy (SEM) images reveal the formation of well defined uniform shaped α-Fe2O3 nanospheres. The transmission electron microscopy (TEM) image shows the porous nature of the α-Fe2O3 nanospheres with an interconnected monocrystallites structure. Swagelok type lithium and sodium batteries were fabricated using the developed porous α-Fe2O3 nanostructures as a negative electrode material. As a negative electrode material in Li-ion cells, the porous α-Fe2O3 nanostructures deliver a second cycle discharge capacity of 1000 mA h g−1 at a 0.1 C rate. At a higher current density of 5 C, the porous α-Fe2O3 nanostructures exhibit a specific capacity of 264 mA h g−1. In the case of Na-ion batteries, the porous α-Fe2O3 nanostructures as negative electrode exhibit a reversible capacity of 300 mA h g−1 with excellent cycleability and coulombic efficiency at 0.1 C up to 100 cycles. The observed excellent electrochemical performance is attributed to the porous nature and uniform shape of the α-Fe2O3 nanostructures that are composed of interconnected monocrystallites. Importantly, the architecture of α-Fe2O3 nanostructures, comprising interconnected monocrystallites could be developed within a short time by a rapid microwave synthesis process and it can be applied to develop different morphological nanostructures for better energy storage device applications.
 Please wait while we load your content...
Please wait while we load your content...

