
Interaction of glycine, lysine, proline and histidine with dipalmitoylphosphatidylcholine lipid bilayers: a theoretical and experimental study†
Rodolfo D. Porasso*a,
Norma M. Aleb,
Facundo Ciocco Aloiac,
Diego Masonec,
Mario G. Del Pópoloc,
Aida Ben Altabefbd,
Andrea Gomez-Zavagliae,
Sonia B. Diazb and
Jorge A. Vila*af
aInstituto de Matemática Aplicada San Luis (IMASL), CONICET, Universidad Nacional de San Luis, Italia 1556, CP5700, Argentina. E-mail: rporasso@unsl.edu.ar; vila@unsl.edu.ar
bInstituto de Química Física, Facultad de Bioquímica, Química y Farmacia, U. N. T., San Lorenzo 456, T4000CAN Tucumán, Argentina
cCONICET, Facultad de Ciencias Exactas y Naturales, Universidad Nacional de Cuyo, Padre Jorge Contreras 1300, CP5500, Mendoza, Argentina
dInstituto de Química del Noroeste (INQUINOA) CONICET, Tucumán, Argentina
eCenter for Research and Development in Food Cryotechnology (CIDCA, CCT-CONICET, La Plata), RA-1900, Argentina
fBaker Laboratory of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14853-1301, USA
First published on 5th May 2015
Abstract
The interaction of unblocked glycine, lysine, proline, and histidine (in their three forms, namely two tautomers and the protonated form) with a dipalmitoylphosphatidylcholine (DPPC) bilayer was assessed using extensive atomistic molecular dynamics simulations. Free energy profiles for the insertion of each amino acid into the lipid bilayer were computed along an appropriated reaction coordinate. The simulation results for glycine in the presence of DPPC were compared with experimental data obtained by Fourier transform infrared spectroscopy. Experimental results predict, in good agreement with simulations, the existence of intermolecular interactions between the DPPC head groups and glycine. Atomistic simulations were further extended to investigate the free energy profiles for lysine, proline and histidine, leading to the following conclusions: (i) lysine free energy profiles computed using a united atom force-field and an analog molecule, where the side-chain is truncated at the β-carbon atom, differ significantly from each other; (ii) the free energy profiles for the three forms of histidine are all very similar, although the charged form interacts mostly with the carbonyl groups of DPPC, while the tautomers interact with the phosphate groups; and (iii) proline does not show a minimum in the free energy profile, pointing to the absence of binding to the membrane lipids. Overall, this work contributes to our general understanding of the various factors affecting the interactions between amino acids and a model cell membrane, and may spur progress in the effort to develop new molecular models to study larger biological systems.
1 Introduction
Most of the experimental evidence regarding the interaction of natural amino acids with lipid membranes is commonly interpreted in terms of the chemical nature of the side-chains.1–8 Hence, a usual approach in computer simulations of these systems is to represent the amino acid as an “analog molecule”, consisting of just the side-chain truncated at the β-carbon atom.9–20 However, the use of the analog molecule approach opens the question of to what extent the amino acid backbone influences its partitioning into a lipid bilayer. This problem is particularly important for residues that are not part of regular secondary structure elements in proteins, such as statistical coil fragments or loop regions, which constitute 50% of all residues in proteins.21 The fact that certain amino acids cannot be studied within the analog molecule approximation, e.g., glycine (Gly) and proline (Pro), only exacerbates the problem. Consequently, the aim of this work is threefold. First, to use Gly, which bears no side chain, as a reference compound for testing the additivity of backbone and side chain transfer free energies in all 20 naturally occurring amino acids (except for Pro). Therefore, atomistic Molecular Dynamics (MD) simulations and Fourier Transform Infrared spectroscopy (FTIR) are employed to investigate the nature of Gly–DPPC interactions at a molecular level. The experimental observations will enable us to assess the capabilities and potential limitations of the force-fields used in this work.Second, to carry out MD simulations for the insertion of unblocked charged lysine into a DPPC bilayer, by using both a united-atom representation of the whole amino acid (Lys+) and the analog molecule approach (Lys+-analog). The results of these simulations, together with those for Gly, will be used to discuss the non-additivity of backbone and side chain transfer free energies and, hence, the accuracy and limitations of the analog molecule model. Although it is clear that aqueous-organic transfer free energies cannot in general be decomposed into molecular fragments' contributions, it is important to quantify non-additive effects for amino acid transfers into lipid bilayers, given that analog molecule models are widely used in biophysical simulations. As stated above, the limitations of these models may be severe when transferring residues which are not part of rigid structural motifs in proteins.
Third, to study the transfer free energy profile for both Pro and histidine (His). The reasons for choosing these two amino acids are the following. Proline, which is an imino rather than an amino acid, does not admit an analog molecule representation. Furthermore, His is special among all the ionizable amino acids because it possesses a pK° = 6.6 and, hence, may be charged or neutral around pH 7.0 where most of the biological processes occur. Moreover, for the neutral form of His two tautomers exist, namely Nδ1-H and Nε2-H, which need to be discussed separately.
The rest of the paper is organized as follows: simulation and experimental methods are detailed in Section 2, results are presented and discussed in Section 3, and conclusions are summarized in Section 4.
2 Methodology
2.1 Experimental
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching bands (νCO) were obtained by Fourier self deconvolution using band width parameters between 18 and 20 cm−1 and a band narrowing factor of 2, as defined by the mathematical software GRAMS/32 Spectral Notebase. Deconvolution was used to determine the position of the bands corresponding to the two populations of carbonyl groups in the gel state.23–25
O stretching bands (νCO) were obtained by Fourier self deconvolution using band width parameters between 18 and 20 cm−1 and a band narrowing factor of 2, as defined by the mathematical software GRAMS/32 Spectral Notebase. Deconvolution was used to determine the position of the bands corresponding to the two populations of carbonyl groups in the gel state.23–252.2 Molecular dynamics simulations
MD simulations were used to investigate the insertion of Gly, Pro, the three forms of His, and charged Lys (both the united atom model and the analog side-chain molecule), into a DPPC lipid bilayer. The unblocked amino acids were modeled with the GROMOS 53a6 force field.26 For the Lys+ analog, the side chain was truncated at the β-carbon atom. In the united atoms representation of GROMOS 53a6, this was achieved by replacing the original methylene group associated to the β-carbon with a united atom methyl group. The charged forms of the amino acids were neutralized by including a counterion (Cl−) in the simulation cell. DPPC was modeled using the force-field proposed by Berger et al.27 combined with the Single Point Charge (SPC) water model.28 Each simulation box contained 64 DPPC molecules (32 lipids per leaflet), approximately 3815 water molecules (full hydration), and one amino acid initially located at the center of the water slab (z = 3.5 nm from the membrane center). The bilayer normal was perpendicular to the x–y plane of the coordinates system.Since the timescale for the spontaneous penetration of the amino acid into the bilayer is large compared to the simulation time, an external force was applied to the amino acid in order to generate initial configurations for the subsequent free energy calculations. A harmonic potential with a force constant of 3000 kJ mol−1 nm−2 was applied to the reaction coordinate, defined as the z-component of the distance vector between the center of mass of the amino acid and the center of mass of the lipid bilayer.18,29,30 The amino acid was thrust into the lipid bilayer at a rate of approximately 7 nm ns−1, and was allowed to move freely on the x–y plane. The Potential of Mean Force (PMF) for the penetration of the amino acid was computed by Umbrella Sampling31 using a set of 36 windows spanning the reaction coordinate interval 0.0–3.5 nm. Each window was let to relax for 10 ns, and then simulated for over 100 ns. Free energy profiles were recovered with the Weighted Histogram Analysis Method (WHAM).32,33 Convergence was assessed by applying WHAM on consecutive trajectory blocks of 20 ns (see Fig. 1–6 in the ESI†).
All simulations were performed with the GROMACS-4.5.5 package,34,35 using a time step of 2 fs. Lennard–Jones interactions were cutoff at 1 nm, and dispersion corrections were applied to energy and pressure in order to account for the pair-potential truncation. Long range electrostatic interactions were evaluated using the particle mesh Ewald method,36,37 with real space interactions cutoff at 1 nm, and reciprocal space interactions computed on a 0.16 nm grid with a fourth-order spline interpolation. At the beginning of each simulation, a steepest descent minimization process was applied to the whole system in order to remove any excess of strain and potential overlaps between neighboring atoms. Production runs were performed in the NPT thermodynamics ensemble, using as a thermostat the velocity rescaling algorithm of Bussi et al.,38 and a weak pressure coupling algorithm for the barostat.39 The pressure was always set to 1 atm and the temperature to 323 K (above the phase transition temperature, 314 K, of DPPC).40 The coupling constants for the thermostat and the barostat were 0.1 ps and 1 ps, respectively.
3 Results and discussion
3.1 FTIR experiments
Gly is one of most abundant amino acids in nature, and is also involved in several biological processes. More importantly, Gly has no lateral chain and makes an appropriate model for investigating the role of the backbone on the interaction of amino acids with lipid bilayers. This is particularly relevant from the point of view of molecular simulations, as the absence of a side-chain allows us to assess the adequacy of the analog molecule approach. Moreover, the comparison between simulations and experiments delimits the scope of the force-field and the computational strategy employed in this work.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DPPC molar ratios. This indicates that the gel phase of DPPC is not altered by the presence of Gly (see Fig. 1 and Table 1). In addition, no significant shifts, within the experimental error ± 1 cm−1, were observed in the symmetric, antisymmetric and bending modes of the methyl and methylene groups of the inner lipid bilayer in the gel phase (measured at 298.2 K), nor in the crystalline liquid phase (measured at 323.2 K). Frequencies and frequency changes at both temperatures are reported in Tables 1 and 2 of the ESI.†
:DPPC molar ratios. This indicates that the gel phase of DPPC is not altered by the presence of Gly (see Fig. 1 and Table 1). In addition, no significant shifts, within the experimental error ± 1 cm−1, were observed in the symmetric, antisymmetric and bending modes of the methyl and methylene groups of the inner lipid bilayer in the gel phase (measured at 298.2 K), nor in the crystalline liquid phase (measured at 323.2 K). Frequencies and frequency changes at both temperatures are reported in Tables 1 and 2 of the ESI.†
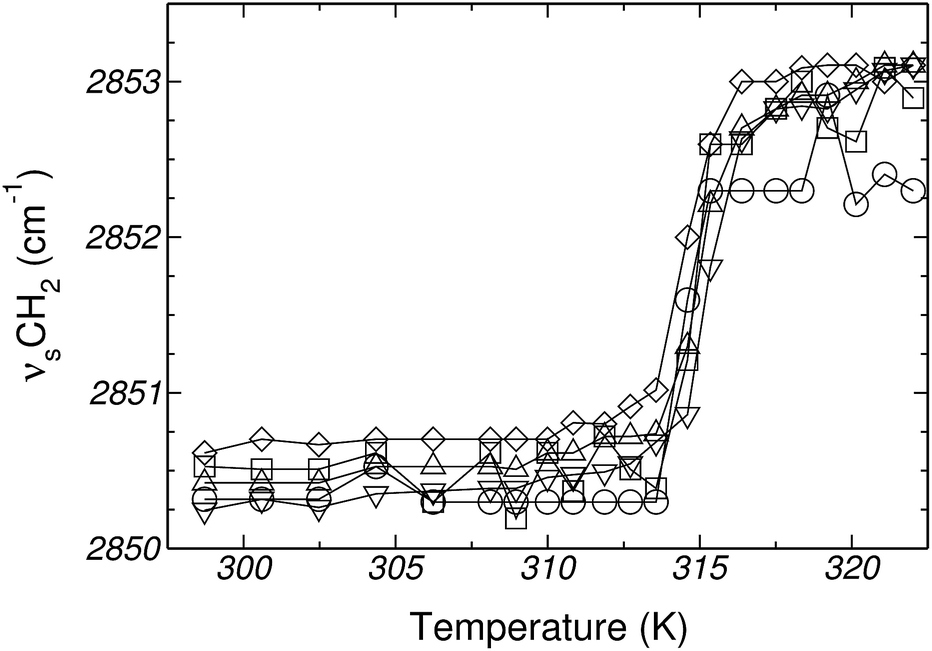 | ||
| Fig. 1 Changes in vibrational frequency of the CH2 symmetric stretching mode in Gly:DPPC (at different molar ratios), as a function of temperature. Gly:DPPC molar ratios: (□) 0.0:1, (○) 0.4:1, (Δ) 0.9:1, (∇) 2.0:1 and (◊) 3.0:1. | ||
:DPPC liposomes (at different molar ratios), both in H2O and D2O
| Molar ratio Gly–DPPC | H2O (K) | D2O (K) |
|---|---|---|
| 0.0:1 |
314.7 | 315.1 |
| 0.4:1 |
314.7 | 314.7 |
| 0.9:1 |
314.7 | 314.7 |
| 2.0:1 |
314.1 | 314.7 |
| 3.0:1 |
313.7 | 313.7 |
| 4.0:1 |
315.6 | 315.6 |
O stretching mode frequency (vCO), and in H2O, to assign the PO2− vibrational mode frequencies.It is well known that the main νCO peak of diacyl lipids can be decomposed into at least two components. One of them corresponds to the H-bonded and the other to the nonbonded (free) conformers of the CO group.44 The higher frequency band component (1740–1742 cm−1) has been assigned to free νCO groups (νCOf), whereas the lower frequency component (∼1728 cm−1) has been attributed to the νCO vibration of H-bonded conformers (νCOb).45 Deconvolution and curve fitting were performed to determine the position and relative contribution of the two carbonyl populations. A large set of spectra and fitting curves are shown in Fig. 1 of the ESI.†
Fig. 2–4 depict the frequency shifts of bonded and free CO groups (νCOb and νCOf, respectively) for Gly:DPPC ratios between 0.0:1 and 4.0:1. Three different temperatures are considered; 298.2 K, corresponding to the gel phase (Fig. 2); the transition temperature 314.2 K (Fig. 3); and 323.2 K corresponding to the liquid crystalline phase (Fig. 4). Numerical values are provided in Table 1 of the ESI.† Below a Gly:DPPC molar ratio of 3.0:1, neither νCOb nor νCOf depict a noticeable shift with respect to the pure lipid at both 298.2 and 323.2 K. However, at the 4.0:1 molar ratio, a smooth shift to lower frequencies is observed for both carbonyl populations (Fig. 2 and 4 and Table 1 of the ESI†).
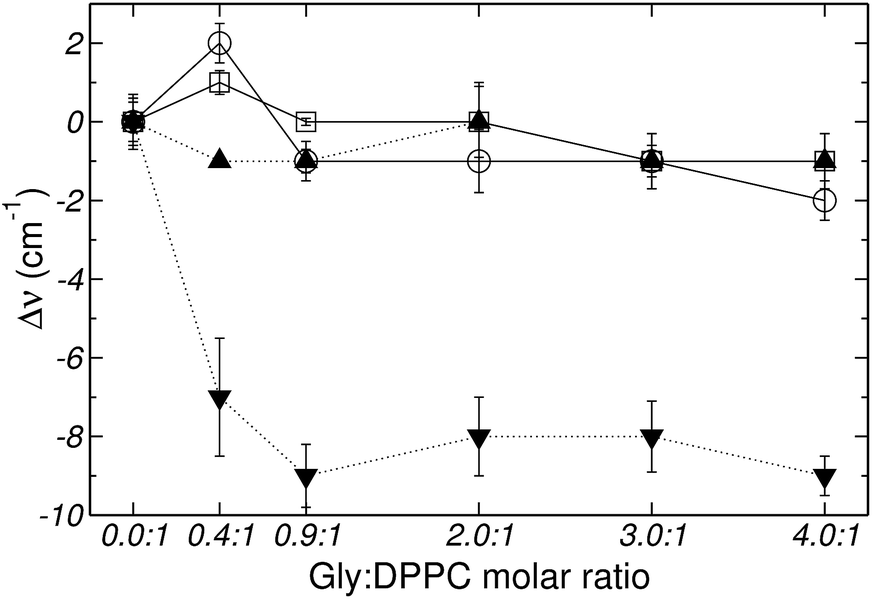 | ||
| Fig. 2 Frequency shifts of: (□) νCOf, (○) νCOb, (▼) vasPO2− and (▲) vsPO2−, stretching vibrational mode as a function of the Gly:DPPC molar ratio at 298.2 K (gel state). | ||
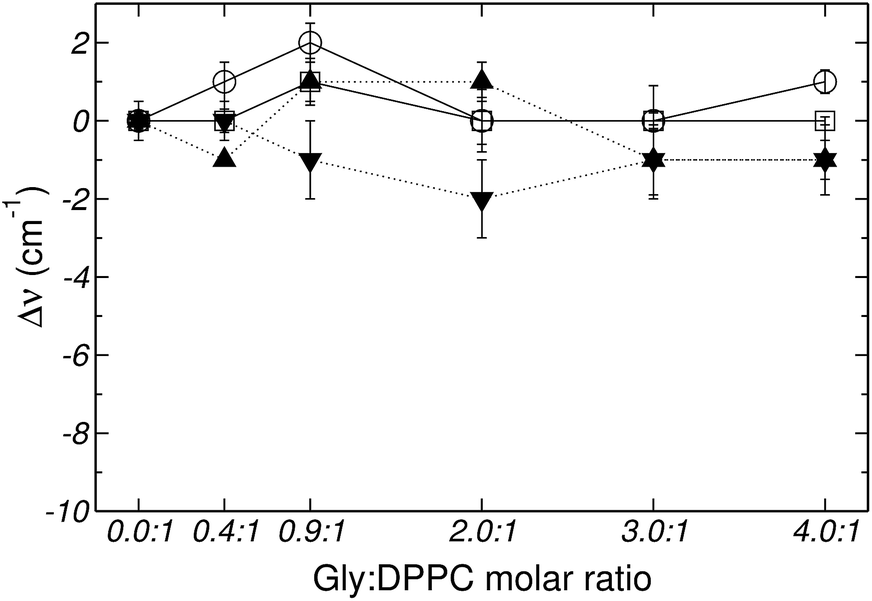 | ||
| Fig. 3 Frequency shifts of: (□) νCOf, (○) νCOb, (▼) vasPO2− and (▲) vsPO2−, stretching vibrational mode as a function of the Gly:DPPC molar ratio at 314.2 K (transition state). | ||
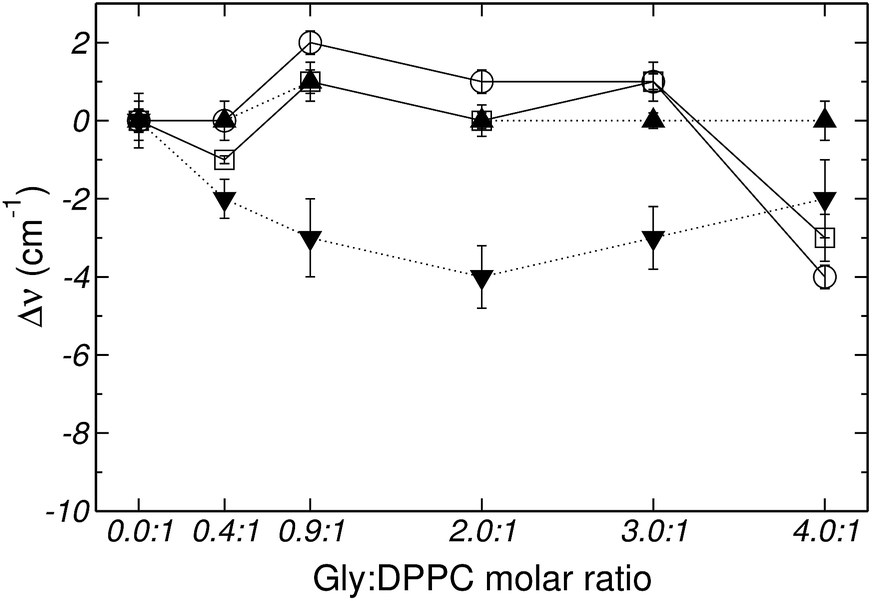 | ||
| Fig. 4 Frequency shifts of: (□) νCOf, (○) νCOb, (▼) vasPO2− and (▲) vsPO2−, stretching vibrational mode as a function of the Gly:DPPC molar ratio at 323.2 K (liquid crystalline state). | ||
Fig. 5 shows the percentage contribution of νCOb and νCOf to the carbonyl stretching mode, taken from Fig. 1 of the ESI,† as a function of the Gly:DPPC molar ratio. In the gel state (298.2 K) the contribution of νCOb is greater than that of νCOf for Gly:DPPC molar ratios between 0.0:1 and 3.0:1. However, the trend inverts at the molar ratio 4.0:1 indicating a saturation of the interphase with Gly molecules (see also Fig. 1aI to VI of ESI†). At the transition temperature (314.2 K) and in the liquid crystalline phase (323.2 K), the contributions of νCOb and νCOf in the pure lipid (Gly:DPPC: 0.0:1) are almost the same, but νCOb becomes clearly dominant when increasing the Gly:DPPC ratio up to 0.9:1 (314.2 K) and 2.0:1 (323.2 K). It must be pointed out that each plot in Fig. 5a, b or c shows the results of experiments carried out at the same temperature (298.2, 314.2 and 323.2 K). In other words, for each set of experiments only the Gly concentration increased and the contribution of water did not change with respect to the pure lipid (Gly:DPPC 0.0:1). Therefore, one could infer that the increase in νCOb contribution indicates the formation of hydrogen bonds between Gly and DPPC.
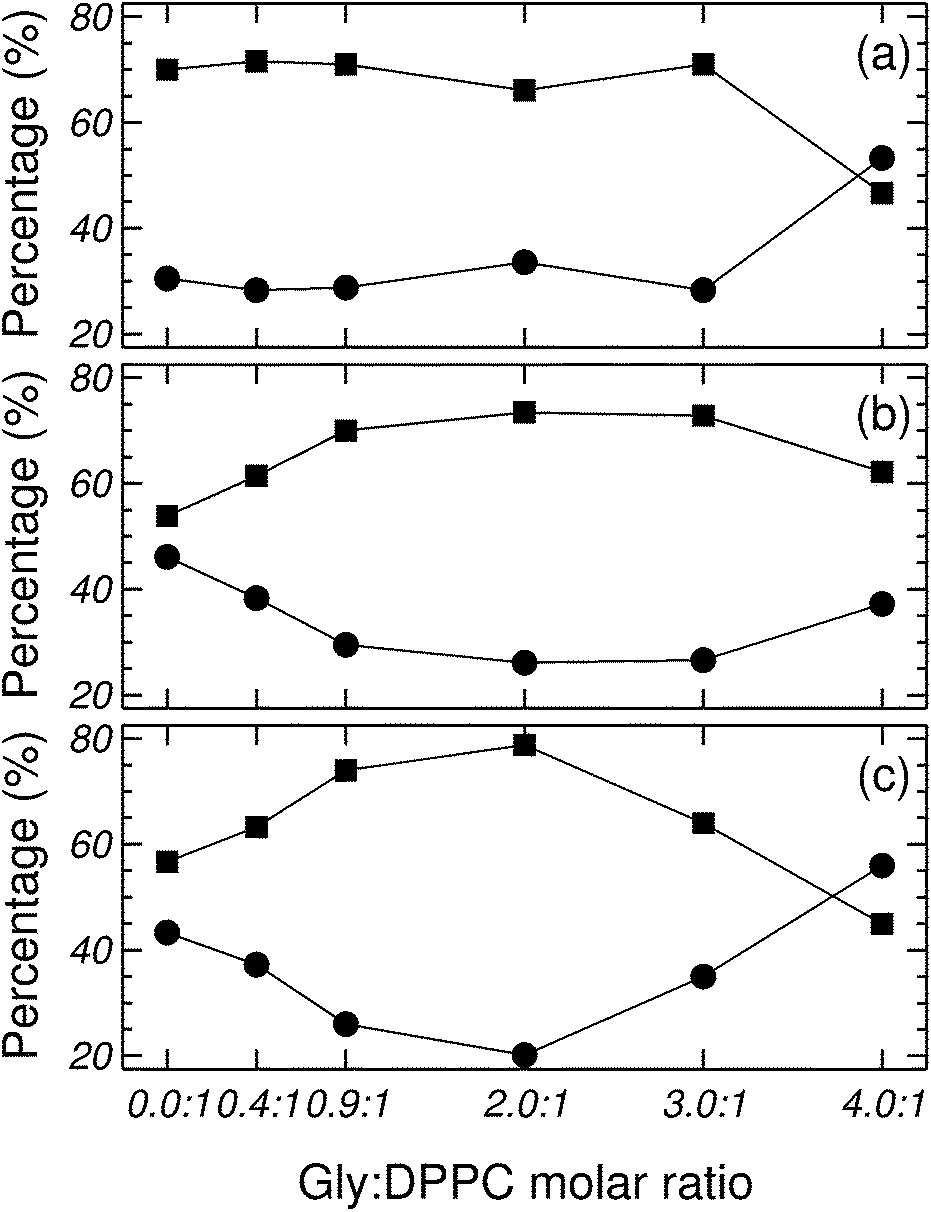 | ||
| Fig. 5 Contribution of νCOf and νCOb to the carbonyl population at different Gly:DPPC molar ratios and at (a) 298, (b) 314.2 and (c) 323.2 K. Symbols indicate: (■) νCOb, and (●) νCOf. | ||
The observations reported in the previous paragraph can be summarized stating that the presence of Gly leads to noticeable changes in νCOf and νCOb (see Fig. 5 and 1 of the ESI†). The evolution of both carbonyl populations was more evident in the fluid state. Assuming that the relative area of a band component is proportional to the respective conformer population, it can be concluded that the populations of CObond and COfree conformers change upon addition of Gly.
The asymmetric stretching mode of the phosphate group (PO2−) shifts to lower frequencies in hydrated lipids.23–25,46,47 This shift has been ascribed to direct H-bonding of water molecules to the charged phosphate groups. Therefore, PO2− has been suggested to act as a sensor of the hydration level of the interphase.23–25,46 Gly had a different quantitative effect on the PO2− stretching bands, depending on whether the bilayer was in the gel (298.2 K) or in the liquid crystalline state (323.2 K) (see Fig. 2, 4 and 6). Independently of the phase state of the membrane, the presence of the amino acid did not show a substantial effect on the PO2− symmetric stretching band. However, the antisymmetric stretching mode showed an important shift (Δv) from −7 to −9 cm−1 in the gel phase, and around −2 to −4 cm−1 in the liquid crystalline state (see Fig. 2 and 4). These observations suggest that in addition to the replacement of water molecules, there is participation of the PO2− groups in the interaction with Gly through H-bonds. As can be inferred from the data reported in Fig. 3, at the transition temperature (314.2 K) and at all Gly molar ratios assayed, there are no significant changes on the antisymmetric and symmetric stretching mode frequencies of the PO2− group.
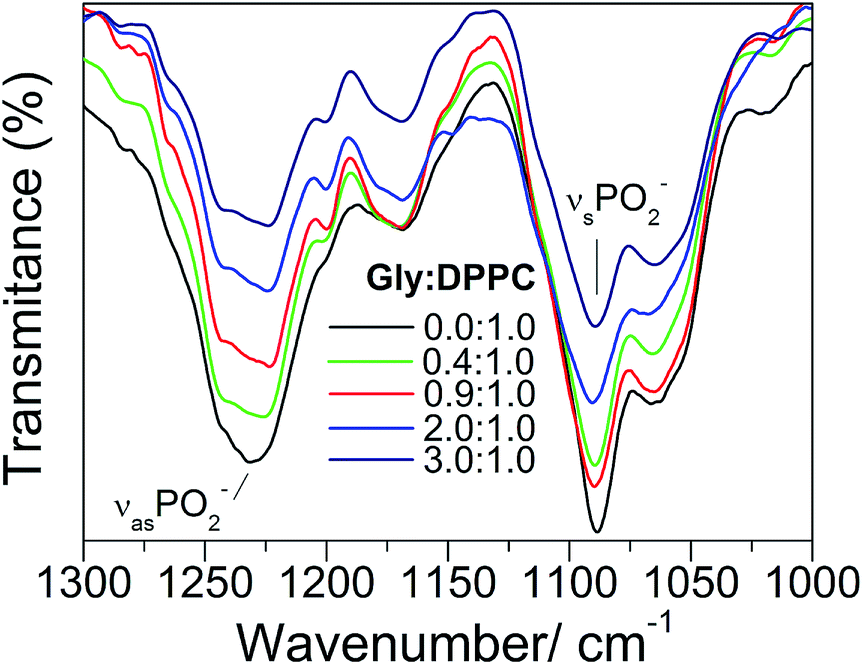 | ||
| Fig. 6 IR spectra for various Gly:DPPC molar ratios in the 1300–1000 cm−1 region. | ||
Overall, the results reported in this section reveal that: (i) the addition of Gly does not alter the fluidity (order of the hydrocarbon chains) of the lipid membrane, and (ii) the presence of specific interactions between the head groups of DPPC and Gly. These observations strongly suggest that the phosphate groups of the lipid membrane form H-bonds with Gly, in replacement of the water molecules, both in the gel and in the liquid crystalline states.
3.2 Molecular dynamics simulations of Gly and Lys
In order to facilitate the description of the free energy profiles, partial local mass density profiles were computed for the simulated bilayer system, and are shown in Fig. 7. Based on these profiles the bilayer is divided into four regions, according to the model used by MacCallum et al.18 Region 1 corresponds to bulk water with a small population of DPPC head groups; region 2 contains most of the charged phosphate and choline atoms; region 3 is a mix between the final portion of the polar head groups and the beginning of the lipids tails; and region 4 includes only the hydrophobic tails.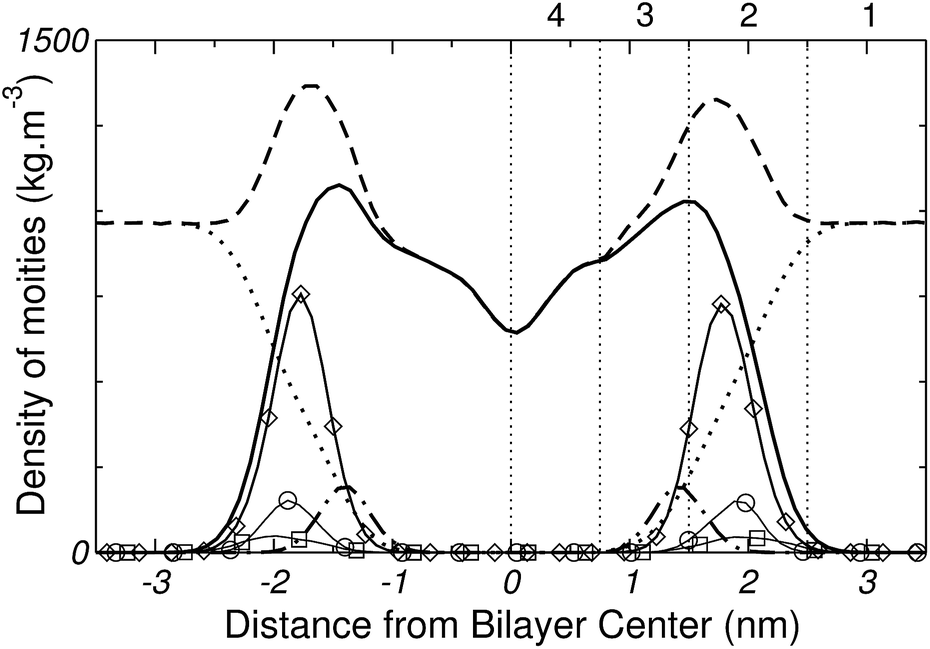 | ||
| Fig. 7 Partial mass density profile of the simulated system. Whole system (---), DPPC (——), water (⋯), lipids' carbonyl groups (– – ), head groups (◊), phosphorous (○) and nitrogen (□). The vertical lines and numbers divide the system into four regions (see text for details). | ||
The free energy profile for inserting Gly into the DPPC bilayer is plotted in Fig. 8. The curve displays a minimum at approximately 1.7 nm from the center of the bilayer and near the core of region 2 (head groups). The free energy gain to bring the amino acid from bulk water to the lipid surface is ∼−40 kJ mol−1. After the minimum, the free energy rises up to ∼50 kJ mol−1, as the amino acid approaches the center of the bilayer. The general features of the free energy profile of Fig. 8 indicate that Gly adsorption on DPPC occurs spontaneously, while its partitioning to the center of the membrane is highly unfavorable. The strong surface binding of the amino acid to the bilayer agrees with the trends discussed in Section 3.1, which suggested that the strongest Gly–DPPC interactions occur at the level of the polar head groups rather than in the membrane core. The adsorption of Gly on DPPC, and its specific interaction with the phosphate groups, is also supported by the simulations and experiments reported in ref. 48.
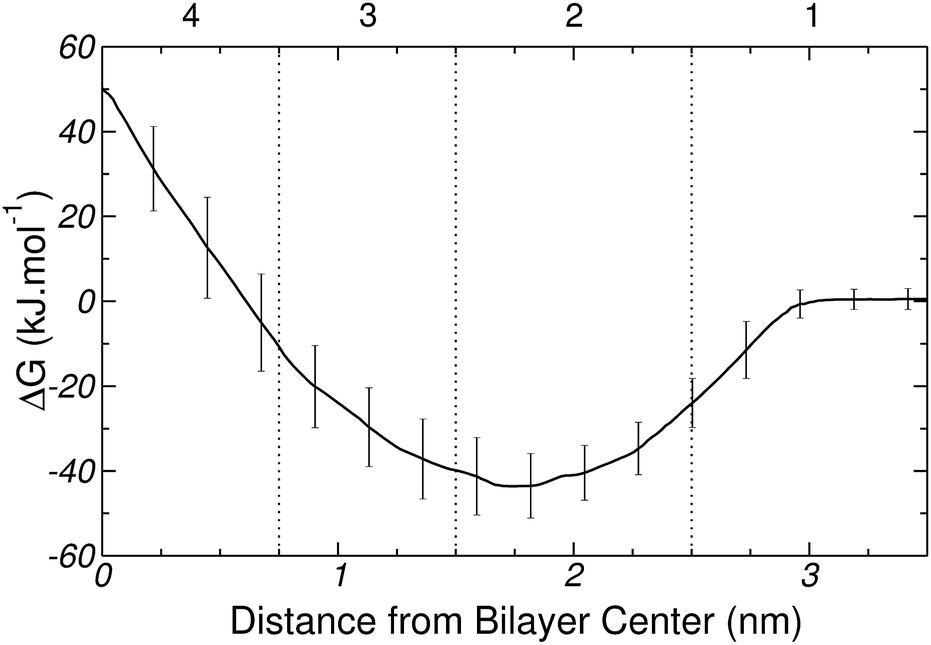 | ||
| Fig. 8 Transfer free energy profile for Gly. Vertical lines divide the system into 4 regions (see Fig. 7). Error bars are standard errors calculated by splitting a 100 ns MD-US trajectory into 5 independent blocks. | ||
In order to characterize changes in the bonding pattern as the amino acid penetrates into the membrane, the number of H-bonds between Gly and water, Gly and DPPC and between DPPC and water was computed as a function of the reaction coordinate z. Fig. 9 demonstrates that the number of Gly–water H-bonds decreases as the amino acid moves into the bilayer, i.e., as it gets into the hydrophobic region of the membrane. On the other hand, Fig. 9 shows that the number Gly–DPPC H-bonds (including bonds to the phosphate and to the carbonyl groups), reaches a maximum in the region of the hydrophilic heads, and decreases slightly as the molecule moves towards the hydrophobic core. It is then clear from Fig. 9 that after traversing region 2 (see Fig. 7), Gly remains partially hydrated and coordinated to a single DPPC head group. This was also confirmed by the inspection of simulation snapshots (see panel A of Fig. 10). For completeness, Fig. 9 shows that the average number of DPPC–water hydrogen bonds changes very little during the insertion of the amino-acid. This can be attributed to the fact that the local perturbation induced by Gly on the DPPC–water interface, is small compared to the total extend of the interface.
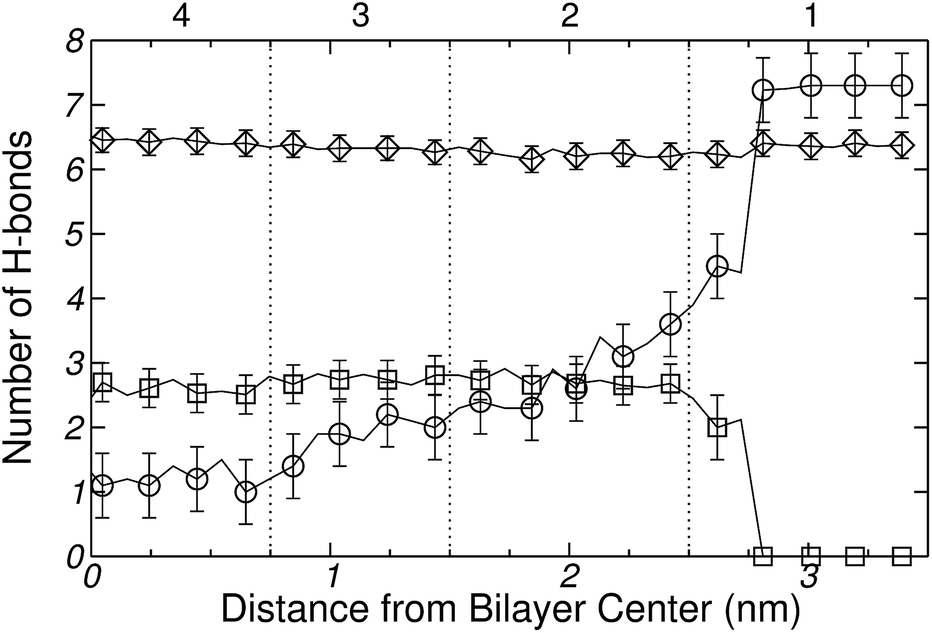 | ||
| Fig. 9 Calculated number of hydrogen bonds formed between: (○) Gly–water (◊) DPPC–water (see text for further explanation) and (□) Gly–DPPC. Error bars calculated from 5 independent simulations. | ||
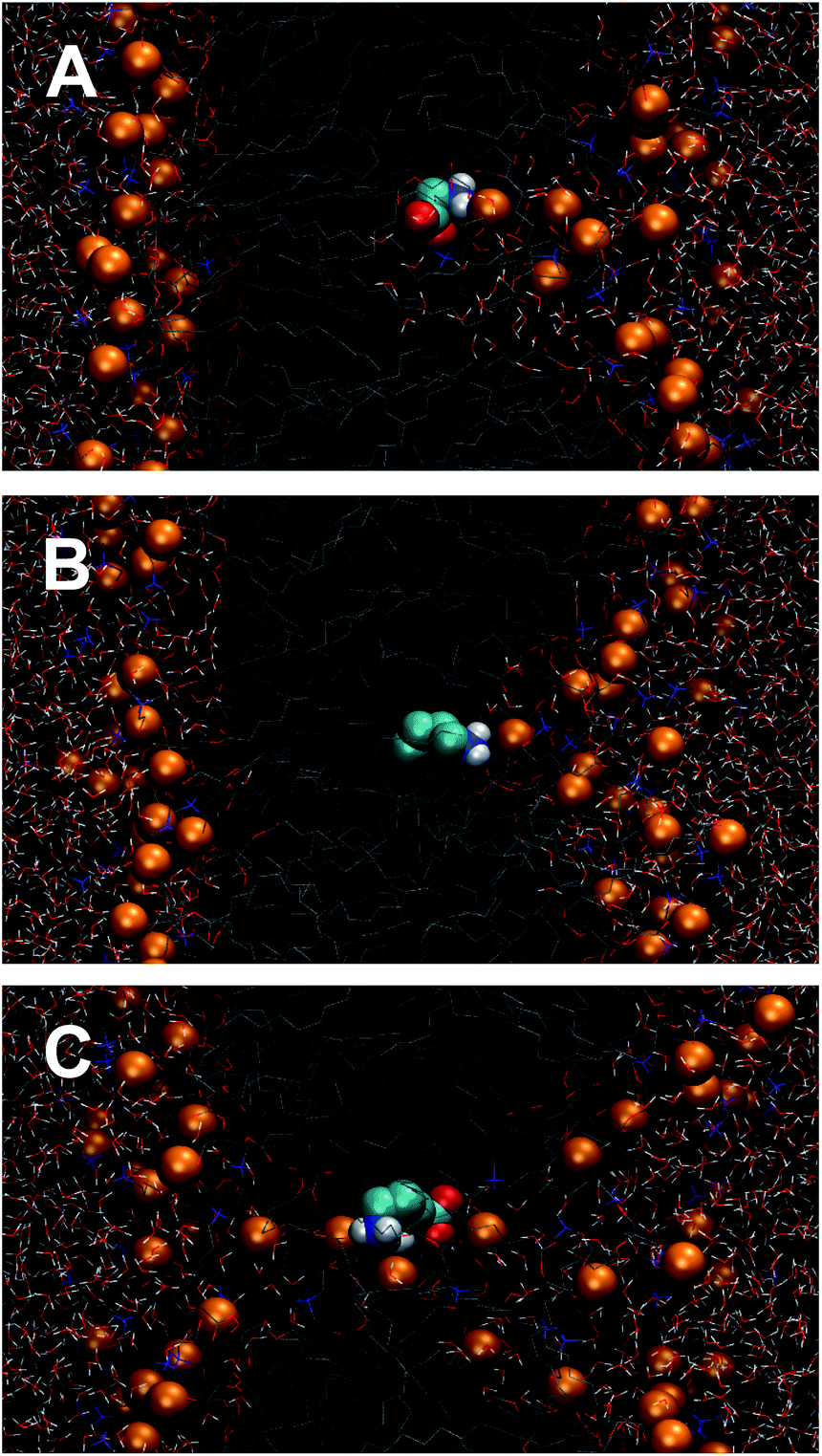 | ||
| Fig. 10 Configurational snapshots extracted from Umbrella Sampling windows located at the center (z = 0) of the DPPC bilayer. (A) Gly, (B) Lys+-analog and (C) whole molecule model of Lys+. The orange beads represent the phosphorous atoms of DPPC. Water molecules are represented by wires. | ||
As stated in Section 1, one of the motivations of the present work is to assess the impact of the analog molecule approach, where amino acids are represented by their side chains, on the thermodynamic work required to bring the molecule from the bulk of the solvent to the surface, or to the center, of the membrane. With this purpose, lysine was chosen as a case study because it possesses a large and flexible side chain, and it also plays important roles in membrane protein activity.20
The free energy profiles for the Lys+-analog and the corresponding whole molecule model, are shown in Fig. 11. As observed, the two free energy curves depict the same general trends (a deep minimum near the membrane surface, and a local maximum at the membrane center), but also significant quantitative differences. In particular, the Lys+-analog (solid curve in Fig. 11) shows almost no free energy change when transferring the molecule from water to the center of the lipid bilayer. Also the free energy minimum (of ∼−57 kJ mol−1) is located within region 3 of the bilayer.
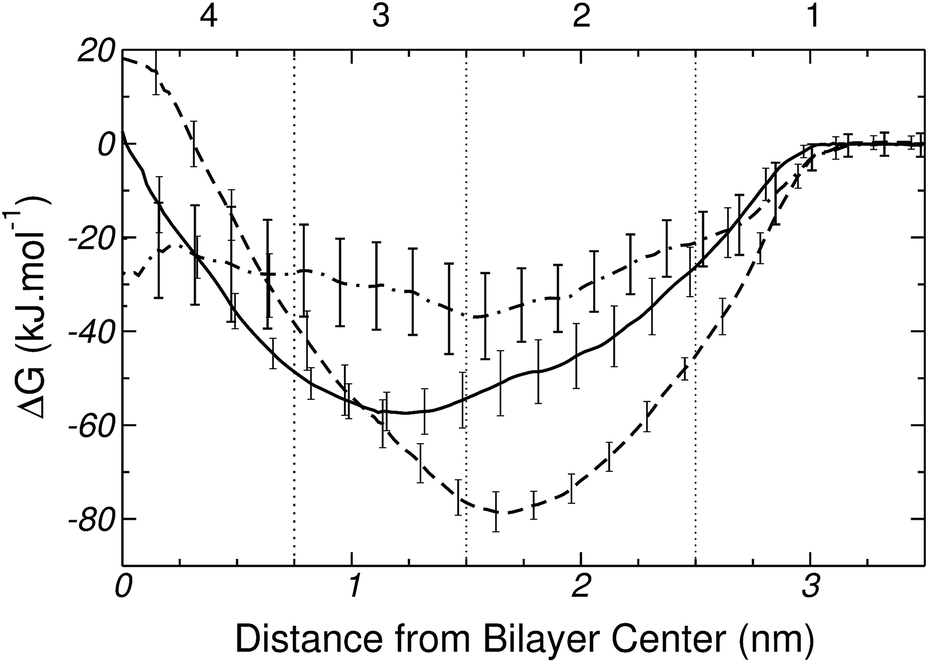 | ||
| Fig. 11 Free energy profiles for Lys+ (---), the Lys+ analog (——), and the difference between the PMFs of Lys+ and Gly (– – ). Vertical lines divide the system into 4 regions (see text for further explanation). Error bars are standard errors computed by splitting a 100 ns MD-US trajectory into 5 independent blocks. | ||
It must be noticed that there is currently a degree of dispersion in the binding energy of amino acid analogs to PC bilayers as predicted by different force-fields. For example, MacCallum et al. have reported a binding energy of around 20 kJ mol−1 for the Lys+-analog on DOPC, based on a combination of Berger's and the OPLS force-field.18 On the other hand, Li et al. have found no adsorption (no minimum in the PMF) of the Lys+-analog on DPPC on the basis of CHARMM. Similar trends have been reported for charged arginine analogs on PC lipids.15,16,18,20 Considering the current discussions in the literature, and the state of the art in computer simulations of protein–lipids systems, we believe that the binding of charged amino acids to zwitterionic membranes is still a topic under scrutiny and debate. The present results provide further elements for judgment.
For the whole-molecule model of Lys+, the maximum in the free energy curve (see dashed line on Fig. 11) occurs at the center of the bilayer (hydrophobic region) and, taking as reference the analog molecule model, the minimum is shifted towards the water–lipid interface. This points to the existence of strong interactions between Lys+ and the polar head groups of DPPC. Fig. 11 also shows a difference of ∼20 kJ mol−1 in the equilibrium adsorption energy, in favor of the whole Lys+ molecule. Such a difference could be traced to a combination factors. The presence of the zwitterionic backbone in the whole-molecule model introduces additional lipid amino acid interactions, and also leads to a larger and tighter hydration shell. Furthermore, the backbone may decrease the conformational freedom of the side-chain and, hence, reduce the entropic contribution to the free energy change. Also, a careful analysis of simulation snapshots reveals that when the molecule is inside the bilayer, the average orientation of the side chain predicted by the two models is different. The isolated chain orients its axis perpendicular to the membrane (x–y) plane, whereas in the whole molecule model the side chain lies on the bilayer plane. Also, panels B and C of Fig. 10 show that the presence of the zwitterionic backbone induces the formation of a water channel across the membrane, which is absent in the isolated chain simulations. Such a trans-membrane defect not only modulates the free energy cost of transferring the molecule to the center of the bilayer, but also conditions the orientation of the amino acid inside the membrane.
Going back to Fig. 11, the dot-dashed line represents the difference between the PMF of the whole Lys+ model and that of Gly. Could the transfer free energy of the amino acid be unambiguously decomposed into a backbone and a side chain contribution, the difference in PMF of Fig. 11 should resemble the profile of the analog molecule model. Clearly, that is not the case, highlighting the non-additivity of backbone and side chain contributions to the transfer free energy. Although analog molecules can be a good approximation to study the membrane insertion of rigid portions of a macromolecule (i.e.: α-helix in proteins), they may be inaccurate to represent the energetic of transferring the most flexible parts (i.e.: loops, turns), which include ∼50% of all amino acidic residues in proteins.
3.3 Molecular dynamics simulations of Pro and His
The transfer free energy profile for Pro is plotted in Fig. 12. The maximum of the free energy curve occurs at the center of the bilayer and is ∼60 kJ mol−1; while the minimum of ∼−9 kJ mol−1, which is quite weak for a polyatomic molecule, is located at the boundary between regions 2 and 3, i.e. close to the carbonyl groups, and at 1.5 nm from the center of the bilayer. Clearly, these results point to an unfavorable interaction between Pro and DPPC.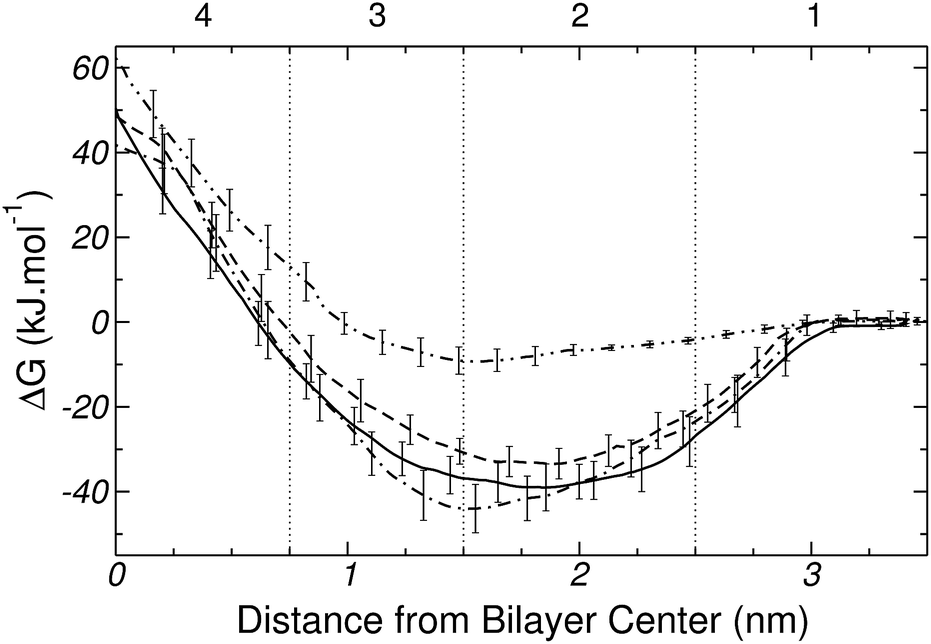 | ||
| Fig. 12 Free energy profiles for Pro (–··–) and the three forms of His, namely, two tautomers [Nδ1-H (——) and Nε2-H (---)], and the protonated form [His+ (– – )]. Error bars are standard errors calculated by splitting a 100 ns MD-US trajectory into 5 independent blocks. | ||
The free energy profiles for the three forms of His are also shown in Fig. 12. Overall the two tautomers (Nδ1-H and Nε2-H) and the ionized form (His+) show very similar trends, with a minimum near the DPPC head groups and a global maximum at the center of the bilayer. Such level of similarity will be explained and discussed in detail in Section 3.4. In the mean time, a few minor differences between the curves of Fig. 12 are worth mentioning. The free energy maxima for the three forms of His have values of ∼50 kJ mol−1, both for Nδ1H and Nε2H, and ∼40 kJ mol−1 for His+. In the neutral tautomers, the depth of the minima differ in only ∼5 kJ mol−1, and are located close to 1.8 nm (near to the phosphate groups). However, for the ionized form (His+), the free energy minimum is ∼5 kJ mol−1 and ∼11 kJ mol−1 deeper than for Nδ1-H and Nε2-H, respectively. Also this minimum is located at the boundary between regions 2 and 3, suggesting specific interactions with the carbonyl groups of DPPC. Overall, our results imply that the three forms of His adsorb spontaneously on the surface of the DPPC bilayer.
3.4 Analysis of hydration/dehydration
Previous simulations of amino acid insertion into lipid bilayers have shown the existence of water molecules trapped into the membrane,15,18,49,50 when the molecule reaches the center of the lipid bilayer. As this effect is also observed in simulations of whole-molecule models, it is worth investigating the impact of the amino acid representation on the amount of hydration water as a function of the reaction coordinate. For example, panels B and C of Fig. 10 already suggest some significant differences in solvation between a whole Lys+ molecule and its analog.Once an atom “X” of a given amino acid is chosen as a reference point (e.g., the carbonyl oxygen of the backbone), the number of hydrating water molecules can be calculated from the radial distribution function,
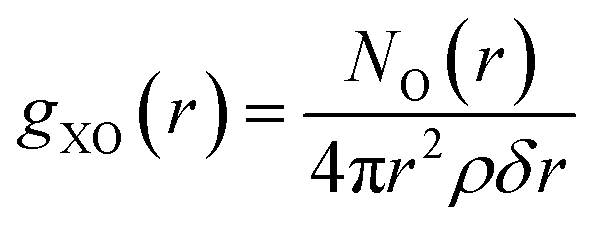 | (1) |
| ΔHN(z) = NOi − NbulkO | (2) |
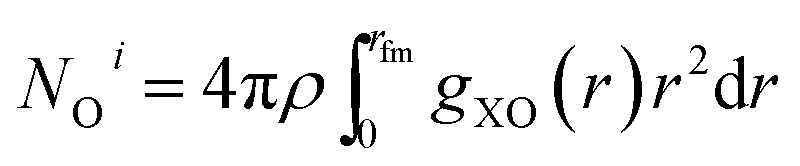 .
.
The dehydration profile of Gly, Lys, Pro and His was determined using eqn (2) for each of the 36 Umbrella Sampling windows, taking as reference (X) the carbonyl oxygen atom of the backbone. The results shown in Fig. 13 indicate that, except for charged His, all the amino acids exhibit a similar dehydration pattern as they are inserted into the bilayer, i.e., each amino acid looses a total of 10–12 water molecules after insertion. In contrast, charged His looses much less hydration water (only 3 water molecules on the average) than the corresponding neutral tautomers. The His+ ion seems to be effectively shielded by a tightly bound layer of hydration water, which could explain the similarity between the free energy profiles of His+ and His reported in Fig. 12.
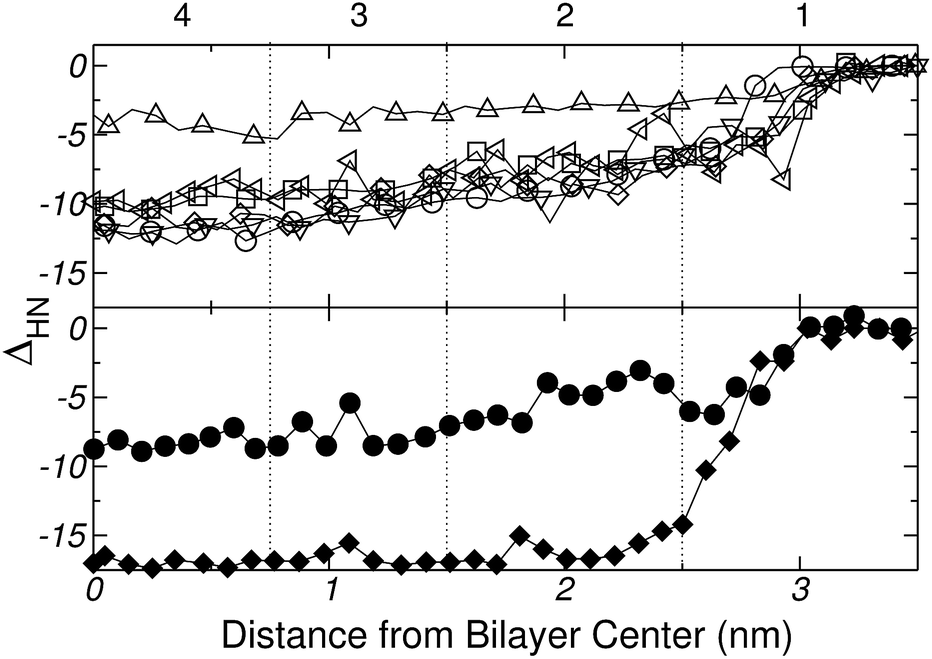 | ||
| Fig. 13 Dehydration (ΔHN) of the amino acids as they are inserted into the lipid bilayer (see text for further explanation). Vertical lines divide the system into 4 regions as in Fig. 7. Upper panel: ΔHN for Gly (○), Lys+ (◁), Pro (∇), His+ (Δ), Nδ1-H (□), and Nε2-H (◊), measured from the carbonyl oxygen atom of the backbone. Bottom panel: ΔHN for the analog (♦) and whole molecule model (●) of Lys+ measured from the nitrogen atom of the lateral chain. | ||
In the case of Gly, Fig. 9 and 13 provide a complementary view of the change in the bonding pattern as the amino acid penetrates into the membrane. Naturally the overall decrease in the number of hydrogen bonds observed in Fig. 9a, is concomitant with the decrease in the number of hydrating water molecules shown in the upper panel Fig. 13. In particular, when Gly reaches the center of the bilayer (see Fig. 7, of the ESI†) it losses 11, but retain (on average) ∼2–3, water molecules; among the retained water molecules, only one forms hydrogen bond with Gly (see Fig. 9a). Simultaneously, Gly forms one hydrogen-bond with the phosphate group of a lipid molecule (see Fig. 9b and 10A).
Finally, the bottom panel of Fig. 13 shows ΔHN(z) for both the whole and the analog molecule model of Lys+, computed from the nitrogen atom of the lateral chain (X = Nchain in eqn (1)). Clearly, the two models lead to significant quantitative differences in the number of solvating water molecules as the amino acid moves towards the center of the bilayer.
3.5 Unbiased simulations of amino acids exclusion from the bilayer centre
In order to test whether the amino-acids could remain trapped in a metastable state when reaching the centre of the bilayer (potential local minima not captured in the free-energy profiles), unbiased MD trajectories were initiated from the top of the free-energy barrier. In all cases simulations were started from configurations that had evolved under Umbrella Sampling for 100 ns. Fig. 14 shows the time evolution of the distance, along the bilayer normal, between the center of mass (COM) of the membrane and the COM of the amino acid, after removing the harmonic restraint. The results are presented in the following order: (a) Gly, (b) Nε2-H, (c) Nδ1-H, (d) His+, (e) Lys+, (f) Lys+-analog and (g) Pro. In all the seven cases the amino acid spontaneously leaves the membrane core, and migrates towards the bilayer surface. This occurs within a time scale of a few tens of nanoseconds. Gly, His (in the three forms), Lys+ and the Lys+-analog end up exploring the minimum of the free energy profiles reported in Sections 3.2 and 3.3. In the special case of Pro, where a weak interaction with the membrane was found, it can be appreciated that the amino acid leaves the bilayer core, moves freely into the solvent, and finally gets in contact with the bilayer surface. The horizontal dashed lines in Fig. 14 represent the average distance to the bilayer centre, once the time series has stabilised. These values are collected in Table 2 (column daz) and compared with the position of the minima of the corresponding free-energy profiles (column dbz). Clearly, within a timescale of at most 50 ns all amino acids reach the thermodynamic equilibrium position.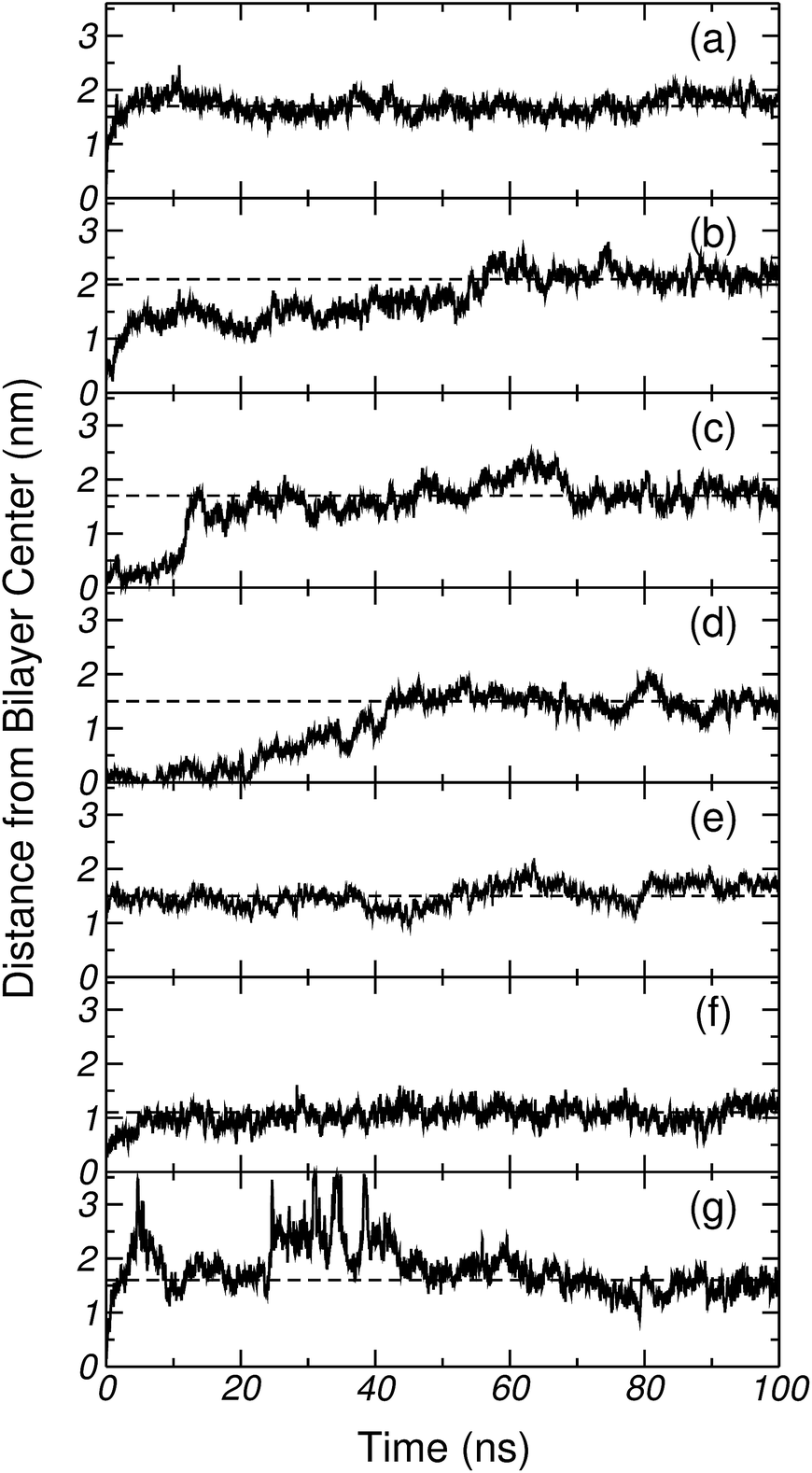 | ||
| Fig. 14 Distance, between the center of mass of DPPC and the corresponding amino acids, along the normal to the bilayer plane. (a) Gly, (b) Nε2-H, (c) Nδ1-H, (d) His+, (e) Lys+, (f) Lys+-analog and (g) Pro. The horizontal dashed lines, represents the average value of such distance after discarding the transient. | ||
| Amino acid | daz (nm) | dbz (nm) |
|---|---|---|
| Gly | 1.7 ± 0.2 | ∼1.7 |
| Nε2-H | 2.1 ± 0.3 | ∼1.9 |
| Nδ1-H | 1.7 ± 0.3 | ∼1.8 |
| His+ | 1.5 ± 0.2 | ∼1.5 |
| Lys+ | 1.5 ± 0.2 | ∼1.6 |
| Lys+-analog | 1.1 ± 0.2 | ∼1.1 |
| Pro | 1.6 ± 0.2 | ∼1.5 |
4 Conclusions
Molecular dynamics simulations and FTIR experiments were used to investigate the interaction of a selected set of amino acids (Gly, Lys, Pro and three forms of His) with a DPPC bilayer. All the amino acids were considered to be in the zwitterionic form. Free energy profiles for the insertion of the amino acids into the membrane were computed by Umbrella Sampling, using as reaction coordinate the z-distance between the center of the bilayer and the amino acid. Given that Gly bears no side chain, it was taken as a reference system to investigate the backbone and side chain contributions to the free energy cost for transferring amino acids from the aqueous phase to the surface and bulk of the lipid membrane. Both simulations and experiments showed that Gly adsorbs spontaneously on the surface of DPPC, forming distinguishable hydrogen-bonds with the lipids' phosphate groups.The analysis of free energy profiles for the insertion of Lys+, computed with a whole molecule model of the amino acid and the commonly used analog molecule approach, showed that Lys+ adsorbs strongly on DPPC and its insertion into the bilayer incurs a high energy penalty. More importantly, the comparison between the two PMFs for Lys and the PMF for Gly (backbone analog) demonstrated that the water–lipid transfer free energy of Lys+ can not be decomposed into additive side chain and backbone contributions. This puts a note of caution on the use of analog molecules when computing the transfer energy of peptides and flexible portions of proteins, such as statistical coil fragments or loop regions, which involve ∼50% of all residues in proteins.21
Finally, Pro and His exhibit their own peculiarities and were discussed separately. Due to its chemical structure, Pro does not admit an analog molecule representation. Our calculations showed that this imino acid only exhibits unfavorable interactions with DPPC. At the same time the free energy profiles for the three forms of histidine resulted to be all very similar, although the charged form interacts mostly with the carbonyl groups of DPPC, while the tautomers do with the phosphate groups. Also, when entering the bilayer, the charged form of His preserves a significantly larger amount of hydration water than the two neutral tautomers.
Acknowledgements
R.D.P., M.G.D.P., A.B.A., A.G.-Z. and J.A.V. are staff members of CONICET (National Research Council, Argentina). F.C.A. and D.M. fellowships are funded by CONICET. The computational component of this work has been financed though the following grants: PIP-112-2011-0100030-CONICET; P-328402 from UNSL, Argentina; PICT-2759 ANPCyT, Argentina. Experimental data has been supported by grants PICT(2011)/0226 and PICT-(2013)/0697, Argentina. The authors acknowledge the Computer Center staff of the Instituto de Matemática Aplicada San Luis for their technical support in carrying out the simulations of this work.References
- R. Wolfenden, L. Andersson, P. M. Cullis and C. C. B. Southgate, Biochemistry, 1981, 20, 849–855 CrossRef CAS.
- J.-L. Fauchère, M. Charton, L. B. Kier, A. Verloop and V. Pliska, Int. J. Pept. Protein Res., 1988, 32, 269–278 CrossRef PubMed.
- A. Radzicka and R. Wolfenden, Biochemistry, 1988, 27, 1664–1670 CrossRef CAS.
- A. Kim and F. C. Szoka Jr, Pharmaceut. Res., 1992, 9, 504–514 CrossRef CAS.
- T. Hessa, H. Kim, K. Bihlmaier, C. Lundin, J. Boekel, H. Andersson, I. Nilsson, S. H. White and G. von Heijne, Nature, 2006, 433, 377–381 CrossRef PubMed.
- G. von Heijne, J. Gen. Physiol., 2007, 129, 353–356 CrossRef CAS PubMed.
- R. Wolfenden, J. Gen. Physiol., 2007, 129, 357–362 CrossRef CAS PubMed.
- S. White, J. Gen. Physiol., 2007, 129, 363–369 CrossRef CAS PubMed.
- H. R. Guy, Biophys. J., 1985, 47, 61–70 CrossRef CAS.
- W. C. Wimley, T. P. Creamer and S. H. White, Biochemistry, 1996, 35, 5109–5124 CrossRef CAS PubMed.
- A. Villa and A. E. Mark, J. Comput. Chem., 2002, 23, 548–553 CrossRef CAS PubMed.
- M. R. Shirts, J. W. Pitera, W. C. Swope and V. S. Pande, J. Chem. Phys., 2003, 119, 5740–5761 CrossRef CAS PubMed.
- W. Gu, S. J. Rahi and V. Helms, J. Phys. Chem. B, 2004, 108, 5806–5814 CrossRef CAS.
- D. Bemporad, J. W. Essex and C. Luttmann, J. Phys. Chem. B, 2004, 108, 4875–4884 CrossRef CAS.
- J. L. MacCallum, W. F. D. Bennet and D. P. Tieleman, J. Gen. Physiol., 2007, 129, 371–377 CrossRef CAS PubMed.
- S. Dorairaj and T. W. Allen, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4943–4948 CrossRef CAS PubMed.
- T. W. Allen, J. Gen. Physiol., 2007, 130, 237–240 CrossRef CAS PubMed.
- J. L. MacCallum, W. F. D. Bennett and D. P. Tieleman, Biophys. J., 2008, 98, 3393–3404 CrossRef PubMed.
- D. Sengupta, J. C. Smith and G. M. Ullmann, Biochim. Biophys. Acta, Biomembr., 2008, 1778, 2234–2243 CrossRef CAS PubMed.
- L. Li, I. Vorobyov and T. W. Allen, J. Phys. Chem. B, 2013, 117, 11906–11920 CrossRef CAS PubMed.
- B. Rost and C. Sander, J. Mol. Biol., 1993, 232, 548–599 CrossRef PubMed.
- A. D. Bangham, M. W. Hill and N. G. A. Miller, Methods in Membrane Biology, Plenum Press, New York, 1974 Search PubMed.
- W. Hübner and A. Blume, Chem. Phys. Lipids, 1998, 96, 99–123 CrossRef.
- S. B. Díaz, F. Amalfa, A. C. Biondi and E. A. Disalvo, Langmuir, 1999, 15, 5179–5182 CrossRef.
- H. H. Mantsch and R. N. McElhaney, Chem. Phys. Lipids, 1991, 57, 213–226 CrossRef CAS.
- C. Oostenbrink, A. Villa, A. E. Mark and W. F. van Gusteren, J. Comput. Chem., 2004, 25, 1656–1676 CrossRef CAS PubMed.
- O. Berger, O. Edholm and F. Jänhing, Biophys. J., 1997, 72, 2002–2013 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren and J. Hermans, in Intermolecular Forces, Reidel, Dordrecht, 1981, p. 331 Search PubMed.
- R. D. Porasso, W. F. D. Bennett, S. D. Oliveira-Costa and J. J. López Cascales, J. Phys. Chem. B, 2009, 113, 9988–9994 CrossRef CAS PubMed.
- J. J. López Cascales, S. D. Oliveira-Costa and R. D. Porasso, J. Chem. Phys., 2011, 135, 135103(7) Search PubMed.
- G. M. Torrie and J. P. Valleau, J. Comput. Phys., 1977, 23, 187–199 CrossRef.
- S. Kumar, D. Bouzida, R. H. Swensen, P. A. Kollman and J. M. Rosemberg, J. Comput. Chem., 1992, 13, 1011–1021 CrossRef CAS PubMed.
- J. S. Hub, B. L. de Groot and D. van der Spoel, J. Chem. Theory Comput., 2010, 6, 3713–3720 CrossRef CAS.
- B. Hess, D. van der Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS PubMed.
- U. Essmann, L. Perea, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS PubMed.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 014101 CrossRef PubMed.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 8, 3684–3690 CrossRef PubMed.
- A. Seelig and J. Seelig, Biochemistry, 1974, 4839–4845 CrossRef CAS.
- H. L. Casal and H. H. Mantsch, Biochim. Biophys. Acta, 1984, 779, 381–401 CrossRef CAS.
- D. G. Cameron, H. L. Casal and H. H. Mantsch, J. Biochem. Biophys. Methods, 1979, 1, 21–36 CrossRef CAS.
- S. H. White and M. C. Wiener, Determination of the structure of fluid lipid bilayer membranes, in Permeability and Stability of Lipid Bilayers, ed. E. A. Disalvo and S. A. Simon, CRC Press, Boca Raton, 1995, pp. 1–19 Search PubMed.
- A. Blume, W. Huebner and G. Messner, Biochemistry, 1988, 27, 8239–8249 CrossRef CAS.
- R. N. A. H. Lewis and R. N. McElhaney, Infrared Spectroscopy of Biomolecules, Wiley-Liss, New York, 1996, p. 159 Search PubMed.
- J. L. Arrondo, F. M. Goñi and J. M. Macarulla, Biochim. Biophys. Acta, 1984, 794, 165–168 CrossRef CAS.
- W. Pohle, C. Selle, H. Fritzsche and H. Binder, Biospectroscopy, 1998, 4, 267–280 CrossRef CAS.
- C. Wang, F. Ye, G. F. Velardez, G. H. Peters and P. Westh, J. Phys. Chem. B, 2011, 115, 196–203 CrossRef CAS PubMed.
- Y. Hu, S. Ou and S. Patel, J. Phys. Chem. B, 2013, 117, 11641–11653 CrossRef CAS PubMed.
- A. C. V. Johansson and E. Lindahl, Biophys. J., 2006, 91, 4450–4463 CrossRef CAS PubMed.
- J. J. López Cascales, J. García de la Torre, S. J. Marrink and H. J. C. Berendsen, J. Chem. Phys., 1996, 104, 2713–2720 CrossRef PubMed.
- J. J. López Cascales, H. J. C. Berendsen and J. García de la Torre, J. Phys. Chem., 1996, 100, 8621–8627 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Convergence criteria of the free energy profiles and experimental frequency values of the symmetric and antisymmetric stretching and bending modes, are provided. See DOI: 10.1039/c5ra03236a |
| This journal is © The Royal Society of Chemistry 2015 |