
Recent advances in f-element separations based on a new method for the production of pentavalent americium in acidic solution
Bruce J. Mincher*a,
Nicholas C. Schmitta,
Brian K. Schuetza,
Thomas C. Sheheeb and
David T. Hobbsb
aIdaho National Laboratory, Idaho Falls, ID 83415, USA. E-mail: bruce.mincher@inl.gov
bSavannah River National Laboratory, Aiken, SC 29808, USA
First published on 11th March 2015
Abstract
The peroxydisulfate anion has long been used for the preparation of hexavalent americium (AmVI) from the normally stable AmIII valence state in mildly acidic solutions. However, there has been no satisfactory means to directly prepare the pentavalent state (AmV) in that medium. Some early literature reports indicated that the peroxydisulfate oxidation was incomplete, and silver ion catalysis in conjunction with peroxydisulfate became accepted as the means to ensure quantitative generation of AmVI. Incomplete oxidation would be expected to leave residual AmIII, or to produce AmV in treated solutions. However, until recently, the use of peroxydisulfate as an AmV reagent has not been reported. Here, parameters influencing the oxidation were investigated, including peroxydisulfate and acid concentration, temperature, duration of oxidative treatment, and the presence of higher concentrations of other redox active metals such as plutonium. Using optimized conditions determined here, quantitative AmV was prepared in an acidic solution and the UV/Vis extinction coefficients of the AmV 513 nm peak were measured over a range of nitric acid concentrations. The utility of AmV for separations from the lanthanides and curium by solvent extraction, organic column chromatography and inorganic ion exchangers was also investigated.
Introduction
Separations within the f-elements are traditionally considered among the most difficult separations in radiochemistry. This is due to the nearly universal preference of the lanthanides and heavy actinides for the trivalent oxidation state in conjunction with their similar ionic radii in aqueous solution, which afford little difference in charge density for separations using complexing agents. Yet a facile separation of americium from the lanthanides and curium is a current research goal in several fuel cycle countries. The selective separation of americium from used nuclear fuel would reduce the long-term radiotoxicity of nuclear waste destined for the repository and allow that americium to be fissioned for energy production. Thus, new methods for the production and quantitation of higher americium oxidation states is a topic of current interest, with potential applications to separations desired in industry. If higher oxidation states of americium were available, they could offer new options for its separation from the trivalent f-series metals.Although AmIII is the common stable oxidation state in aqueous solutions, higher oxidation states have long been known. The first reported preparation of AmVI was by Asprey et al., in 1951.1–3 They used an excess of solid ammonium peroxydisulfate, (NH4)2S2O8, added to 0.2 M nitric, perchloric or sulfuric acid solutions containing 0.002–0.035 M AmIII. At 85 °C, the pink AmIII color was replaced by a straw-yellow color, and a sharp UV/Vis absorbance peak was reported at 992 nm, with another minor peak at 666 nm. No evidence of AmIII or AmV absorbance was reported. Soon afterward, Ward and Welch reported that the yield of AmVI was only 80% in 0.2 M HNO3.4 They recommended the addition of silver nitrate to ensure quantitative oxidation. The use of silver-catalyzed S2O82− rapidly became the standard method for the production of AmVI for the analytical-scale separation of americium from lanthanides and curium.5–7
Recently, Burns et al. reported that the use of S2O82− in the absence of silver produced only AmV in 0.01 M HNO3 at 80 °C.8 This finding is fortuitous because AmV is more stable in acidic solution than is AmVI and therefore potentially more reliable for separations. Yet options for the preparation of AmV in acidic solution have traditionally been limited to precipitation of the carbonate followed by re-dissolution in acid,9 or the use of sodium bismuthate at elevated temperature.10–12 Use of the carbonate would be tedious to applications which rely on nitric acid processing. And although it is reliable for AmV production the bismuthate oxidation suffers for trivalent metal separations because it introduces BiIII into treated solution.12 Thus, although past reports vary as to its efficacy, an S2O82−-based method for the preparation of AmV directly in acidic solution would be useful. Here we have further investigated the possibility of AmV production using S2O82− and explored its application to separations from curium and the lanthanides.
Experimental
Experiments were performed at the Idaho National Laboratory (INL) and the Savannah River National Laboratory (SRNL). Peroxydisulfate (S2O82−), as either the ammonium (INL work) or sodium salts (SRNL work) (Sigma-Aldrich), and calcium hypochlorite (Fisher Scientific) were reagent grade chemicals and used as received. Concentrated nitric acid was obtained from Fisher Scientific as Optima Trace Metal Grade. Americium, plutonium and curium were obtained from stocks on hand at either laboratory. All solutions were prepared using ultrapure water produced by a MilliQ Element System.To determine the effect of selected oxidation conditions, solutions of 243AmIII were heated to 80–100 °C for durations between 10–60 min, and at varying HNO3 and (NH3)2S2O8 concentrations, following which they were cooled for 5 minutes in an ice bath. The cooled solutions were then filtered through a 0.1 μm Teflon luer-lock filter and injected into a 100 cm liquid waveguide capillary cell (LWCC, World Precision Instruments) and the UV/Vis spectra were recorded using a Cary 50 spectrophotometer (Agilent Technologies). The absorbance of AmIII was monitored at 503 nm, AmV at 513 nm, and AmVI at 666 nm.
The extinction coefficients for AmV (ε513) were also measured using the LWCC/Cary 50 UV/Vis system. The total americium concentration of the stock solution was determined by gamma-ray counting at the 243Am 74.7 keV line. The stock was then diluted in the proper nitric acid concentration and the AmV extinction coefficients were determined under conditions previously found to prepare quantitatively the pentavalent oxidation state. The extinction coefficients were calculated using Beer's law, for concentration ranges with a linear response.
Solvent extraction experiments were performed on americium solutions using the solvent formulation commonly referred to as TRUEX (TRansUranic EXtraction) consisting of 0.2 M octylphenyl-N,N-diisobutylcarbamoylmethylphosphine oxide (CMPO) and 1.1 M tributylphosphate (TBP) in dodecane. It was originally designed as a group separations solvent for the actinides and lanthanides.13 The solvent was washed twice with dilute sodium carbonate to remove possible acidic impurities such as the degradation products of the neutral organophosphorous compounds, and then pre-equilibrated with the appropriate nitric acid concentration for 30 min prior to the actual solvent extraction contacts. The room temperature solvent extraction contact consisted of an equal volume, 15 s hand shake with an aqueous nitric acid solution containing 243Am, followed by a 1 min centrifugation to disengage the phases. Aliquots of each phase were then gamma-counted and the extraction efficiency was reported as the distribution ratio DAm, calculated as the ratio of the activity in the organic phase over that in the aqueous phase. The distribution ratios of 139Ce, 154Eu and 244Cm were determined similarly, except that the curium activity was measured by liquid scintillation counting.
Column separation trials for curium and americium were performed using 2 mL, 50–100 mesh Eichrom TRU resin, (Darien, IL, USA) which contains CMPO/TBP as the ligand. Solutions of 0.01 M HNO3, containing americium and curium were oxidized with 1 M (NH3)2S2O8 at 80–100 °C for 30 min, to ensure AmV. These were cooled to room temperature and the acidity was adjusted to 3 M in HNO3. Then, 250 μL of sample was loaded dropwise. The final concentrations were 1.1 × 10−6 M 243Am and 1.9 × 10−9 M 244Cm. Nominally pentavalent americium was then eluted with 5 mL of 3 M HNO3, followed by elution of CmIII with 5 mL of 0.01 M HNO3. The post elution metal concentrations were determined by alpha spectroscopy, after precipitation as the fluoride on neodymium.
Batch contact ion-exchange experiments were also carried out with two sodium titanate materials; monosodium titanate (MST) obtained from the Optima Chemical Company (Douglas, GA) and SrTreat® obtained from Fortum Engineering Ltd (Finland). Oxidation to AmV was performed in 0.01 M HNO3 using 0.55 M Na2S2O8 at 80–100 °C for 20 min. These experiments were performed before optimized conditions for AmV generation had been identified. Therefore, 1 mL of a 14 mM solution of Ca(OCl)2 was also added to 2.5 mL of sample to reduce any produced AmVI to AmV. The ion-exchange materials were prepared by suspension in water and the pH was adjusted to 3. These were then centrifuged and the excess water removed from the solids. The americium-containing solution was next added to the titanate ion-exchanger, shaken to suspend the solids, and placed on a rotisserie to mix for 24 hours at ambient laboratory temperature (ca. 20 °C). Aliquots of the suspension were periodically taken, filtered through a 0.1 μm Teflon luer-lock filter, and analyzed by alpha spectroscopy to determine the americium and curium activities.
Results and discussion
The UV/Vis absorbance spectrum of a 0.1 M HNO3 solution containing a mixture of americium oxidation states at 4 × 10−5 M total americium is shown in Fig. 1. The spectrum was collected using the 100 cm LWCC cell.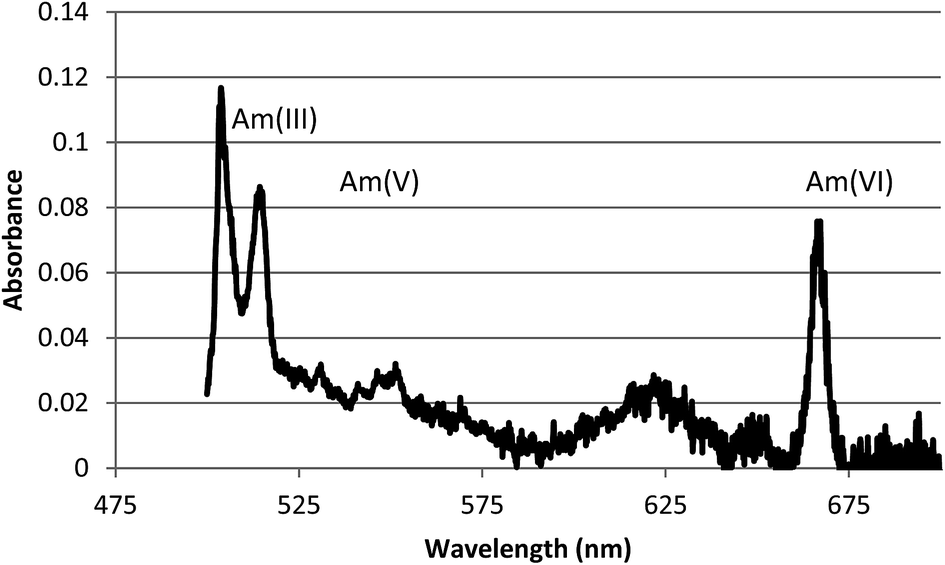 | ||
| Fig. 1 The UV/Vis absorbance of an 0.1 M HNO3 solution containing AmIII (503 nm), AmV (513 nm), and AmVI (666 nm). | ||
Using the absorbance recorded at these wavelengths it was found that oxidation using 0.3 M S2O82− in 0.1 M HNO3 at 80–100 °C yielded a mixture of AmV and AmVI. Oxidation to AmVI continued after cooling for another 5–10 min, following which reduction to AmV commenced, with AmV continuing to grow in for the duration of the experiment. These results are shown in Fig. 2. The concentration decrease for AmVI appears to be linear with respect to time. This zero order reduction with respect to AmVI concentration has been previously reported.14,15
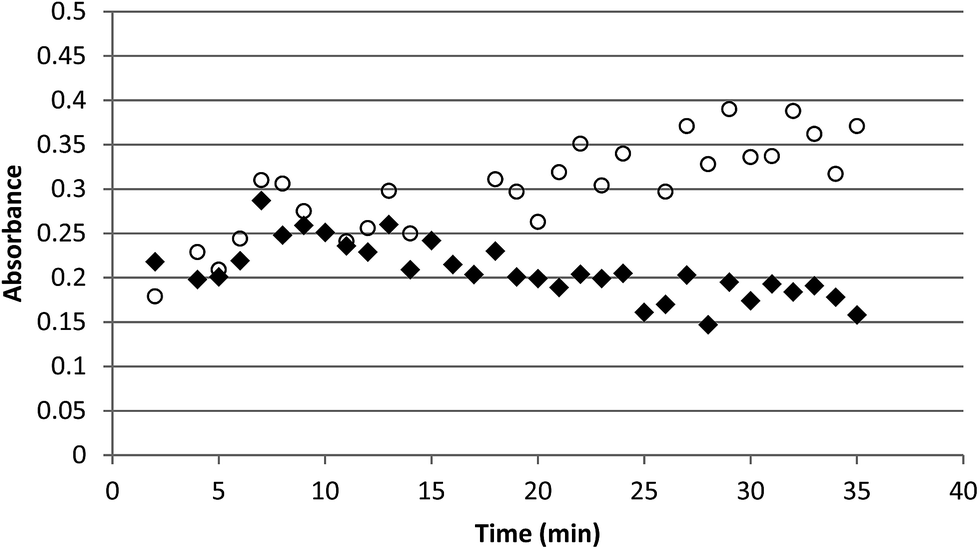 | ||
| Fig. 2 The oxidation of 2.8 × 10−3 M americium using 0.3 M (NH3)2S2O8 in 0.1 M HNO3. Note the continued production of AmVI after the solution cooled (solid diamonds), followed by the decrease in AmVI and increase in AmV absorbance (open circles). | ||
Maintaining this temperature and acid concentration, the effect of the S2O82− concentration on the yield of AmV was measured for 30 min reaction times. At the lowest concentration investigated (0.025 M), a high yield of AmV was achieved, with the balance remaining as AmIII. For 0.05 M S2O82− a mixture of all three oxidation states was obtained. At still higher S2O82− concentrations, the yield of AmVI increased, being ∼60% at 0.3 M S2O82− and being essentially quantitative at 0.5 M S2O82−. Pentavalent americium began to be again produced at higher concentrations and its yield was quantitative at 0.7 M S2O82−.
The preferential yield of AmV at higher S2O82− concentrations is probably due to the reduction of any produced AmVI by H2O2. In acidic solution, protonated S2O82− decays16–18 according to eqn (1)–(3):
| HS2O8− → SO4 + HSO4− | (1) |
| SO4 + H2O → H2SO5 | (2) |
| H2SO5 + H2O → H2O2 + H2SO4 | (3) |
The produced H2O2 of eqn (3) is a reducing agent relative to AmVI, with a reported acid dependent rate constant of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k = (4.592 ± 0.007) − [(0.12 ± 0.01)log[H+].19
k = (4.592 ± 0.007) − [(0.12 ± 0.01)log[H+].19
Based on these results, an optimized concentration of approximately 1 M S2O82− is recommended for the quantitative preparation of AmV in 0.1 M HNO3. The produced AmV was stable for more than 20 h in this solution. It should be noted that Kamoshida and Fukasawa prepared AmVI quantitatively using 1.0 M S2O82− in 1 M HNO3, however; this required the addition of 0.01 M silver as a catalyst.20
It is expected that H2O2 would also be reducing toward AmV. However, the higher stability of AmV in nitric acid solution may indicate that the reaction proceeds with slow kinetics. Therefore we also investigated the effect of varying nitric acid concentrations on the yield of AmV using 1 M S2O82−. As expected,5–7,18,21 the AmV yield rapidly decreased with increasing acidity, consistent with reduction by produced H2O2. Lower yields of AmV were accompanied by increasing amounts of AmIII. At 0.5 M HNO3, only AmIII was detected. The absorbance measured at the AmV 513 nm peak for 1 M S2O82− oxidations at various HNO3 concentrations is shown in Fig. 3.
 | ||
| Fig. 3 The absorbance of the AmV 513 nm peak measured in the LWCC/Cary 50 for 1 M (NH3)2S2O8 oxidations of 3.5 × 10−5 M Am, performed at various HNO3 concentrations. | ||
When 1 M S2O82− oxidation in 0.1 M HNO3 was performed at 80–100 °C for durations of 10–45 min, AmV was produced quantitatively. For a 60 min treatment, traces of AmIII were detected. This may indicate that at long oxidation times the decomposition of S2O82− becomes sufficient to produce enough H2O2 to begin reducing AmV.
Finally, Fig. 4 shows that the oxidation of americium in the presence of a much higher 242Pu concentration was not adversely affected. The slightly higher nitric acid concentration of 0.15 M HNO3 while still using 1.0 M S2O82− was chosen to mitigate the possibility of plutonium hydrolysis, while remaining within the range at which americium oxidation was shown above to be efficient. Oxidation of 3.0 × 10−5 M americium to AmV in the presence of 6.1 × 10−4 M plutonium was not adversely affected. It can be seen in Fig. 4 that AmV (513 nm) was produced nearly quantitatively when the plutonium concentration was 20× higher than the americium concentration. There is only about a 1% contribution from AmIII (503 nm). Although plutonium was probably oxidized to PuVI under these conditions, it did not interfere with the oxidation of the lower concentration of americium.
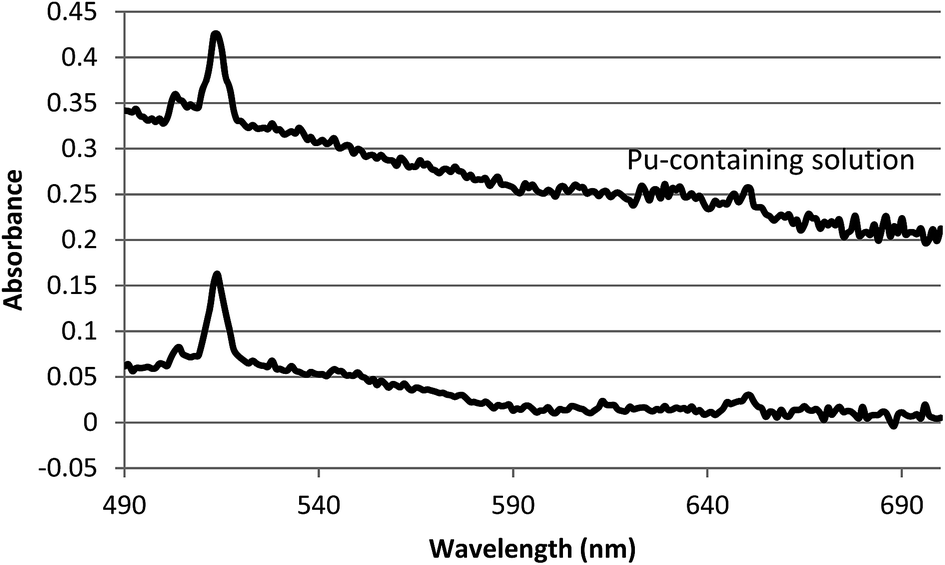 | ||
| Fig. 4 The UV/Vis absorbance spectra of 3.0 × 10−5 M americium following oxidation with 1 M (NH3)2S2O8 in 0.15 M HNO3. Spectra collected using the LWCC/Cary 50. The baselines are off-set for ease of comparison. | ||
The ability to quantitatively prepare AmV in HNO3 solution provided the opportunity to evaluate the molar extinction coefficients for the 513 nm AmV peak over a range of acid concentrations. The values measured are summarized in Table 1. The ε513 for AmV is constant over a wide range of nitric acid concentrations, being decreased only slightly at 6.5 M HNO3. Since AmV exists as the low charge density AmO2+ ion, it is not surprising that AmV does not form significant nitrate complexes, unlike AmIII, PuIII, PuIV and PuVI, which have molar extinction coefficients significantly affected by nitric acid concentration.22,23 A mean value of 39.9 ± 1.6 M−1 cm−1 is reported here for ε513 over the acid concentration range 0.01–1.0 M.
| [HNO3] (M) | ε513 |
|---|---|
| 0.01 | 41.2 ± 1.24 |
| 0.1 | 38.1 ± 2.80 |
| 1.0 | 40.3 ± 1.62 |
| 6.5 | 36.4 ± 0.06 |
Previous values of the molar extinction coefficient of the AmV 513 nm peak have been reported ranging from 40–48 M−1 cm−1 in 0.1 M H2SO4 and HClO4.24
The neutral organophosphorous compound CMPO is used in the proposed TRUEX nuclear fuel cycle process as a group actinide and lanthanide complexing agent. It strongly complexes these elements in their III, IV and VI oxidation states.13 It is not efficient for the extraction of pentavalent actinides and the resulting DAmV should be much lower than DLnIII and DCmIII.12 Therefore, CMPO solvent extraction experiments using S2O82− oxidation were performed to evaluate the potential for the separation of AmV from these elements. The TRUEX separation of AmV from cerium, europium and curium is shown in Fig. 5, following adjustment of the acid concentrations after the initial oxidation. A separation factor of 60 was obtained for the lanthanides and curium from americium, for extractions from 3 M HNO3. The behavior of the lanthanides and curium was identical, indicating that cerium was not oxidized to CeIV, probably due to the production of H2O2.
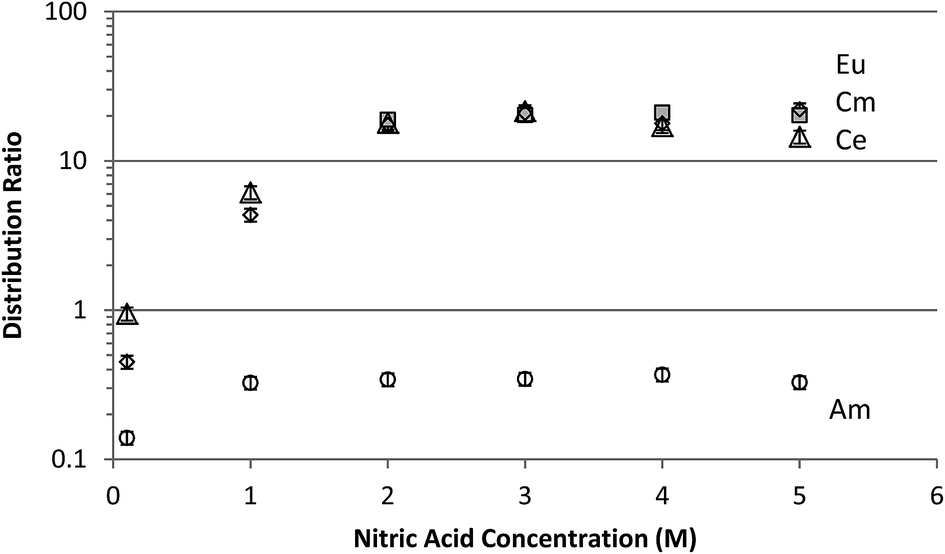 | ||
| Fig. 5 The TRUEX solvent extraction of nominally AmV (circles), EuIII (diamonds) CeIII (triangles), and CmIII (shaded boxes) from 1 M (NH3)2S2O8-oxidized solution, after adjustment of the nitric acid concentration to the indicated values. The average mass balance for the data was 1.03 ± 0.03. Error bars shown are ±10%. | ||
Shehee et al. reported that when S2O82− americium oxidation was performed in neutral perchlorate solution in the presence of lanthanides, a solid precipitate appeared.25 It was identified as a double sulfate of the lanthanide and sodium, with the source of sulfate apparently being decomposition of S2O82− (see eqn (3)). Although no precipitates occurred in our acidic solutions, Kamoshida and Fukasawa20 reported that sulfate produced from the decomposition of S2O82− impeded the extraction of AmVI by TBP from acidic solution. However, as can be seen in Fig. 5, the presence of unavoidably high concentrations of sulfate following decomposition of the S2O82− did not hinder efficient extraction of the trivalent metals using the TRUEX solvent.
The partitioning of americium and curium was next attempted using commercially available TRU columns (Eichrom, Darien, IL). These columns are commonly used in actinide analytical separations. They also contain CMPO/TBP as the ligand and are thus expected to illustrate behavior similar to that shown above in the TRUEX solvent extraction experiments. An S2O82−-treated solution containing 1990 Bq mL−1 (1.1 × 10−6 M) 243Am and 1396 Bq mL−1 (1.9 × 10−9 M) 244Cm was adjusted to 3 M in HNO3 and loaded onto the columns. Nominally pentavalent americium was eluted with 3 M HNO3 followed by a curium elution with 0.01 M HNO3. The resulting eluents were analyzed by alpha spectroscopy. The mean results for triplicate trials are shown in Table 2.
| Feed | Am activity | 1990 |
| Cm activity | 1396 | |
| Am eluent | Am activity | 962 ± 52 |
| Cm activity | 0.82 ± 1.03 | |
| Cm eluent | Am activity | 389 ± 60 |
| Cm activity | 1410 ± 81 |
It can be seen in Table 2 that the curium was well partitioned from the americium fraction with a DF ∼ 1700; however, the americium yield was low at only ∼50%. This low yield is attributable to reduction of AmV on the column, and the failure to elute the resulting AmIII by the 3 M HNO3 eluent intended to collect AmV. The curium was quantitatively collected by elution with dilute acid, although this fraction was contaminated with an amount of americium representing 20% of the initial concentration, probably eluting as AmIII. The total americium recovery was <70%, indicating that some americium was still sorbed to the column after the curium elution. The reduction of AmV on the columns is probably due to reduction by the polymeric resin substrate or the organic ligands themselves, since AmV is stable in nitric acid. The short contact times (15 s) with these same ligands when used in solvent extraction contacts may have mitigated this reduction.10
Elimination of the organic ligands was next used to mitigate the reduction of AmV during column separations. To demonstrate the partitioning of oxidized americium from curium by inorganic ion exchange, batch contact tests were performed with two sodium titanate materials. Sodium titanates feature a layered structure in which sodium ions are sandwiched between the anionic titania layers. The coordination geometry of the TiIV is a distorted octahedral environment. These materials exhibit an affinity for a wide range of metal ions in both alkaline and acidic solutions. One of the sodium titanates, monosodium titanate (NaHTi2O5·xH2O) (MST) is used at the Savannah River Site to remove strontium and actinides from alkaline high-level waste solutions.26,27 Another layered titanate, sodium nonatitanate (Na4Ti9O20·xH2O), is available commercially as SrTreat® and has been used to treat cooling basin waters and cleaning solutions.28,29
For these experiments, a dilute nitric acid solution (pH 3) containing 1 × 10−6 M americium and 2.5 × 10 −6 M curium was treated with 0.55 M Na2S2O8 to oxidize americium to AmV. Calcium hypochlorite, which reduces AmVI to AmV, was added to ensure the pentavalent oxidation state.8 The SrTreat® and MST were then added to separate aliquots of the oxidized solution at a phase ratio of 100 mL g−1. After a 24 hour contact time, the test mixtures were filtered and the filtrate analyzed for the americium and curium activities. Distribution values, Kd, for were calculated from the solution activities and are reported in Table 3. Separation factors (SF), calculated as the ratio of the respective Kd values, are also presented.
| Material | Kd Am | Kd Cm | SF Cm/Am |
|---|---|---|---|
| MST | 167 ± 17 | 18700 ± 933 |
112 ± 11 |
| SrTreat® | 109 ± 5 | 15400 ± 769 |
142 ± 7 |
The Kd values measured for CmIII were 18700 and 15400 mL g−1, which are consistent with values reported for AmIII and LnIII ions in dilute nitric acid and indicate strong interaction between the Am3+ cation and the sodium titanate ion-exchanger. The Kd values for the oxidized americium were much lower, 167 and 109 mL g−1, for MST and SrTreat®, respectively. The low americium Kd values are consistent with reduced interaction due to the lower charge density of the monovalent americyl species, AmO2+, and the sodium titanate ion exchanger. Consequently, the SFs for the sodium titanate materials measured 112 ± 11 for MST and 142 ± 7 for SrTreat® and are sufficiently high to afford very good separation of americium and curium.
Conclusions
Traditionally used to oxidize americium to AmVI, the peroxydisulfate anion was found to be useful for the production of AmV in dilute acidic solution. Appropriate conditions for the production of the pentavalent oxidation state are 1 M S2O82−, 0.1 M HNO3, 80–100 °C for 10–45 min. Quantitative yields of AmV under these conditions were verified spectroscopically, and the molar extinction coefficient ε513 is reported to be 39.9 ± 1.6 M−1 cm−1 over the HNO3 concentration range 0.01–1.0 M. The presence of a 20-fold higher concentration of plutonium did not interfere with this oxidation. The utility of AmV in separations from the trivalent f-elements was shown using both solvent extraction and ion exchange techniques. With short-term contact times, solvent extraction using CMPO/TBP solutions provided good separation of americium from cerium, europium and curium. Longer contact with the same ligands supported on a polymeric resin resulted in some reduction of americium and the degradation of separations efficiency. However, the use of inorganic, titanate-based ion exchangers resulted in very high separation factors for americium from curium. Thus, the ability to prepare AmV in high yields directly in the acidic media most often of interest offers exciting new opportunities in separations design among the f-elements.Acknowledgements
The INL and SRNL work was performed under the US DOE Office of Nuclear Energy Fuel Cycle R&D Sigma Team for Minor Actinide Separations Program under Idaho Operations Contract DE-AC07-05ID14517 and Savannah River Operations Contract DE-AC09-08SR22470.References
- L. B. Asprey, S. E. Stephanou and R. A. Penneman, A new oxidation state of americium, Am(VI). Report AECU-927, United States Atomic Energy Commission, 1950.
- L. B. Asprey, S. E. Stephanou and R. A. Penneman, J. Am. Chem. Soc., 1950, 72, 1425 CrossRef CAS.
- L. B. Asprey, S. E. Stephanou and R. A. Penneman, J. Am. Chem. Soc., 1951, 73, 5715 CrossRef CAS.
- M. Ward and G. A. Welch, J. Chem. Soc., 1954, 4038 CAS.
- H. P. Holcomb, Anal. Chem., 1954, 36, 2329 CrossRef.
- F. L. Moore, Anal. Chem., 1971, 43, 487 CrossRef CAS.
- F. D. Hindman, Anal. Chem., 1986, 58, 1238 CrossRef CAS.
- J. D. Burns, T. C. Shehee, A. Clearfield and D. T. Hobbs, Anal. Chem., 2012, 84, 6930 CrossRef CAS PubMed.
- J. S. Coleman, T. K. Keenan, L. H. Jones, W. T. Carnal and R. A. Penneman, Inorg. Chem., 1963, 2, 58 CrossRef CAS.
- B. J. Mincher, L. R. Martin and N. C. Schmitt, Inorg. Chem., 2008, 47, 6984 CrossRef CAS PubMed.
- B. J. Mincher, L. R. Martin and N. C. Schmitt, Solvent Extr. Ion Exch., 2012, 30, 445 CrossRef CAS.
- B. J. Mincher, N. C. Schmitt and M. E. Case, Solvent Extr. Ion Exch., 2011, 29, 247 CrossRef CAS.
- E. P. Horwitz, D. G. Kalina, H. Diamond, G. F. Vandegrift and W. W. Schulz, Solvent Extr. Ion Exch., 1985, 3, 7 Search PubMed.
- L. B. Asprey and S. E. Stephanou, The autoreduction of Am(VI) and Am(V) in dilute acid. Report AECU-924, United States Atomic Energy Commission, 1950.
- G. R. Hall and T. L. Markin, J. Inorg. Nucl. Chem., 1957, 4, 296 CrossRef CAS.
- E. J. Behrman and J. O. Edwards, Rev. Inorg. Chem., 1980, 2, 179 CAS.
- D. A. House, Chem. Rev., 1962, 62, 185 CrossRef CAS.
- I. M. Kolthoff and I. K. Miller, J. Am. Chem. Soc., 1951, 73, 3055 CrossRef CAS.
- M. Woods Sr, A. Cain and J. C. Sullivan, J. Inorg. Nucl. Chem., 1974, 36, 2605 CrossRef.
- M. Kamoshida and T. Fukasawa, J. Nucl. Sci. Technol., 1996, 33, 403 CrossRef CAS.
- F. Moore, Anal. Chem., 1963, 35, 715 CrossRef CAS.
- W. W. Schulz, The chemistry of americium, Report TID-26971, ERDA Critical Review Series, Technical Information Center, Energy Research and Development Administration, Oak Ridge, TN, USA, 1976.
- M. H. Lee, Y. J. Park and W. H. Kim, J. Radioanal. Nucl. Chem., 2007, 273, 375 CrossRef CAS.
- W. H. Runde and B. J. Mincher, Chem. Rev., 2011, 111, 5723 CrossRef CAS PubMed.
- T. Shehee, L. R. Martin, P. R. Zalupski and K. L. Nash, Sep. Sci. Technol., 2010, 45, 1743 CrossRef CAS.
- D. T. Hobbs, M. J. Barnes, R. L. Pulmano, K. M. Marshall, T. B. Edwards, M. G. Bronikowski and S. D. Fink, Sep. Sci. Technol., 2005, 40, 3093 CrossRef CAS.
- T. B. Peters, M. J. Barnes, D. T. Hobbs, D. D. Walker, F. F. Fondeur, M. A. Norato and S. D. Fink, Sep. Sci. Technol., 2006, 41, 2409 CrossRef CAS.
- A. Clearfield and J. Lehto, J. Solid State Chem., 1988, 73, 98 CrossRef CAS.
- J. Lehto, L. Brodkin and R. Harjula, SrTreat – a highly effective ion exchanger for the removal of radioactive strontium from nuclear waste solutions, 6th International Conference on Radioactive Waste Management and Environmental Remediation, Singapore, 1996, pp. 245–248 Search PubMed.
| This journal is © The Royal Society of Chemistry 2015 |