
Exploring and exploiting the influence of the compression mechanism on the transport properties of CrF3
A. H. Reshak*ab
aNew Technologies – Research Centre, University of West Bohemia, Univerzitni 8, 306 14 Pilsen, Czech Republic. E-mail: maalidph@yahoo.com; Tel: +420 777729583
bCenter of Excellence Geopolymer and Green Technology, School of Material Engineering, University Malaysia Perlis, 01007 Kangar, Perlis, Malaysia
First published on 7th May 2015
Abstract
We have explored the influence of the compression mechanism and the temperature on the transport properties of CrF3. The charge carrier concentration decreases with increasing temperature while it decreases with increasing pressure up to 1.84 GPa; then, after a notable increase in the carrier concentration occurs to reach the maximum value when the pressure rises up to 3.43 GPa, further increases in pressure cause a significant reduction in the carrier concentration to reach the lowest value at 5.43 GPa. Raising the pressure up to 8.56 GPa causes a huge increase in the carrier concentration. An increase in pressure up to 9.12 GPa leads to a drop in the carrier concentration again to the lowest value which was already obtained at 5.43 GPa. Therefore, the pressure value 3.43 GPa is the optimal pressure to gain the highest carrier concentration. It has been found that an electrical conductivity reduction occurs by increasing the temperature within the investigated pressure range. The electrical conductivity significantly increases with increasing pressure, suggesting that the CrF3 could be a good thermoelectric material. Increasing the temperature within the investigated pressure range causes significant increases (almost linearly) in the Seebeck coefficient. The Seebeck coefficient represents n-/p-type conductions. The electronic thermal conductivity ke increases linearly with increasing temperature, and at high temperature one can see that increasing the pressure causes a small reduction in ke. It has been noted that the power factor is zero till 160 K, then above this temperature the power factor increases rapidly with increasing temperature. At high temperatures a reduction in power factor occurs with increasing pressure.
1. Introduction
In 1957 Jack and Maitland1 investigated the metal trifluorides CrF3, afterwards further investigation was carried out by Knox2 in 1960. It has been reported that the metal trifluorides CrF3, which crystallize in the rhombohedral structure, are composed of corner-sharing CrF6 octahedra (structurally related to the perovskite type materials). Each CrF6 octahedron consists of two F–F distances. In late 2004 Jorgensen et al.3 investigated the structural properties of CrF3 using time-of-flight neutron powder diffraction; the investigation was done under ambient pressure, then the pressure increased gradually up to 9.12 GPa. It has been noted that within the investigated pressure range there is a volume reduction during the rotation of the CrF6 octahedra, with a decrease in the bond angles of Cr–F–Cr from 144.80(7)° to 133.9(4)° and a small octahedral strain was found with increasing the pressure. Later on several investigations have been done on materials having the VF3-type structure with the formula MX3, including CrF3.4–11From the above, it is clear that there is a lack of information regarding the transport properties of CrF3. Therefore, we thought it worthwhile to perform comprehensive theoretical calculation based on the density functional theory to investigate the thermoelectric properties of CrF3 at different pressures and temperatures. Calculations are performed using full potential method within BoltzTraP code12 to ascertain the effect of the pressure and temperatures on the electronic structure and hence on the thermoelectric properties. It has been proven that the first-principles calculation is a strong and useful tool to predict the crystal structure and its properties related to the electron configuration of a material before its synthesis.13–16
2. Details of calculations
Based on the experimental work of Jorgensen et al.3, the influence of the pressure on the electronic structure and hence the transport properties of CrF3 were comprehensively investigated by means of density functional theory (DFT). CrF3 crystallizes in hexagonal structure with space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c. We have used the experimental lattice parameters reported by Jorgensen et al.3 at different pressures starting from ambient pressure up to 9.12 GPa. Seeking meaningful comparison, the current theoretical investigations were performed at the same pressure values which were investigated experimentally.3 The experimental lattice constants3 were optimized using Perdew, Burke and Ernzerhof generalized gradient approximation (PBE-GGA).22 The experimental atomic positions3 were relaxed by minimizing the forces acting on each atom; we assume that the structure is totally relaxed when the forces on each atom reach values less than 1 mRy a.u.−1. The optimization of the atomic positions was achieved using PBE-GGA.
c. We have used the experimental lattice parameters reported by Jorgensen et al.3 at different pressures starting from ambient pressure up to 9.12 GPa. Seeking meaningful comparison, the current theoretical investigations were performed at the same pressure values which were investigated experimentally.3 The experimental lattice constants3 were optimized using Perdew, Burke and Ernzerhof generalized gradient approximation (PBE-GGA).22 The experimental atomic positions3 were relaxed by minimizing the forces acting on each atom; we assume that the structure is totally relaxed when the forces on each atom reach values less than 1 mRy a.u.−1. The optimization of the atomic positions was achieved using PBE-GGA.
The optimized lattice parameters were shown in Table 1 in comparison with the experimental data,3 good agreement was found. It is clear that the a-lattice constant decreases by around 7.2% with increasing pressure, whereas the c-lattice constant increases by around 0.17% to reach its maximum at 3.43 GPa, which is associated with the small distortion in the CrF6 octahedra, then above 3.43 GPa it decreases with increasing the pressure, this observation agrees well with the experimental finding.3
| P (GPa) | a (Å) | c (Å) | Cr 6(b) | F 18(e) | MT-Cr | MT-F |
|---|---|---|---|---|---|---|
| 0.0001 (exp.) | 4.9863(2) | 13.2142(7) | (0.0 0.0 0.0) | (0.6153(3) 0.0 0.25) | ||
| 0.0001 (opt.) | 4.9908 | 13.2211 | (0.0 0.0 0.0) | (0.6161 0.0 0.25) | 1.83 | 1.74 |
| 1.01 (exp.) | 4.9106(2) | 13.2286(9) | (0.0 0.0 0.0) | (0.6271(3) 0.0 0.25) | ||
| 1.01 (opt.) | 4.9191 | 13.2361 | (0.0 0.0 0.0) | (0.6282 0.0 0.25) | 1.83 | 1.74 |
| 1.84 (exp.) | 4.8593(2) | 13.2329(9) | (0.0 0.0 0.0) | (0.6338(3) 0.0 0.25) | ||
| 1.84 (opt.) | 4.8612 | 13.2401 | (0.0 0.0 0.0) | (0.6344 0.0 0.25) | 1.83 | 1.74 |
| 3.43 (exp.) | 4.7860(3) | 13.237(1) | (0.0 0.0 0.0) | (0.6419(6) 0.0 0.25) | ||
| 3.43 (opt.) | 4.7896 | 13.2410 | (0.0 0.0 0.0) | (0.6401 0.0 0.25) | 1.82 | 1.73 |
| 5.43 (exp.) | 4.7189(2) | 13.219(1) | (0.0 0.0 0.0) | (0.6524(8) 0.0 0.25) | ||
| 5.43 (opt.) | 4.7201 | 13.2271 | (0.0 0.0 0.0) | (0.6587 0.0 0.25) | 1.82 | 1.73 |
| 7.46 (exp.) | 4.6639(2) | 13.192(1) | (0.0 0.0 0.0) | (0.658(1) 0.0 0.25) | ||
| 7.46 (opt.) | 4.6701 | 13.2011 | (0.0 0.0 0.0) | (0.6572 0.0 0.25) | 1.82 | 1.73 |
| 8.56 (exp.) | 4.6382(2) | 13.172(1) | (0.0 0.0 0.0) | (0.660(1) 0.0 0.25) | ||
| 8.56 (opt.) | 4.6401 | 13.1842 | (0.0 0.0 0.0) | (0.6620 0.0 0.25) | 1.81 | 1.72 |
| 9.12 (exp.) | 4.6265(3) | 13.163(1) | (0.0 0.0 0.0) | (0.659(1) 0.0 0.25) | ||
| 9.12 (opt.) | 4.6313 | 13.1741 | (0.0 0.0 0.0) | (0.6571 0.0 0.25) | 1.81 | 1.72 |
We have used the semi-classical Boltzmann theory as incorporated in the BoltzTraP code12 to calculate the transport properties of CrF3 at different temperatures and pressures starting from ambient pressure up to 9.12 GPa to ascertain the influence of temperatures and the compression mechanism on the transport properties. It has been confirmed that no phase transition occurs within the investigated pressure range.3 The carrier concentration (n), Seebeck coefficient (S), electrical conductivity (σ/τ), electronic thermal conductivity (κe/τ), and the electronic power factor (S2σ/τ) as a function of temperature at a certain value of chemical potential (see Table 2) as well as a function of chemical potential at three constant temperatures (300, 600 and 900 K) for different pressure values are theoretically investigated. The constant relaxation time approximation and the rigid band approximation are used in the calculations.12 The relaxation time is taken to be direction independent and isotropic.17
| P (GPa) | n (e uc−1) @50 K | n (e uc−1) @900 K | σ/τ × 1020 (Ω m s)−1 @50 K | σ/τ × 1020 (Ω m s)−1 @900 K | K × 1014 (W m−1 K−1 s−1) @50 K | K × 1015 (W m−1 K−1 s−1) @900 K | Chemical potential (μ) (eV) |
|---|---|---|---|---|---|---|---|
| 0.0001 | 0.029 | −0.021 | 0.748 | 0.843 | 1.044 | 2.075 | 0.21634 |
| 1.01 | −0.038 | −0.078 | 0.829 | 0.843 | 1.044 | 1.962 | 0.23553 |
| 1.84 | 0.009 | −0.064 | 0.901 | 0.862 | 1.0869 | 1.941 | 0.24986 |
| 3.43 | 0.078 | −0.021 | 0.954 | 0.904 | 1.192 | 1.925 | 0.27526 |
| 5.43 | −0.053 | −0.124 | 1.03 | 0.941 | 1.263 | 1.910 | 0.29745 |
| 7.46 | −0.030 | −0.107 | 1.09 | 0.983 | 1.327 | 1.941 | 0.31746 |
| 8.56 | 0.056 | −0.038 | 1.12 | 1.016 | 1.348 | 1.987 | 0.33141 |
| 9.12 | −0.053 | −0.122 | 1.13 | 1.016 | 1.404 | 2.024 | 0.33828 |
The BoltzTraP code depends on a well tested smoothed Fourier interpolation to obtain an analytical expression of bands. This is based on the fact that the electrons contributing to the transport are in a narrow energy range due to the delta-function-like Fermi broadening. For such a narrow energy range the relaxation time is nearly the same for the electrons. The accuracy of this method has previously been well tested, and the method actually turns out to be a good approximation.18 The temperature dependence of the energy band structure is ignored. The formulae of the transport coefficients as a function of temperature and chemical potential are given somewhere else.12,19 To gain high thermoelectric efficiency, it is important that the material possesses high electrical conductivity, a large Seebeck coefficient and low thermal conductivity.20 The electronic band structures are calculated using full potential linear augmented plane wave (FPLAPW+lo) method in a scalar relativistic version as embodied in the WIEN2k code.21 The exchange–correlation (XC) potential is solved using generalized gradient approximation (PBE-GGA)22 which is based on exchange–correlation energy optimization to calculate the total energy. The unit cell was divided into two regions; the spherical harmonic expansion was used inside the non-overlapping spheres of muffin-tin radius (RMT) and the plane wave basis set was chosen in the interstitial region (IR) of the unit cell. The RMT for Cr and F were chosen in such a way that the spheres did not overlap, these values are listed in Table 1. In order to get the total energy convergence, the basis functions in the IR were expanded up to RMT × Kmax = 7.0 and inside the atomic spheres for the wave function. The maximum value of l was taken as lmax = 10, while the charge density is Fourier expanded up to Gmax = 12 (a.u)−1. Self-consistency was obtained using 300  points in the irreducible Brillouin zone (IBZ). The self-consistent calculations are converged since the total energy of the system is stable within 0.00001 Ry. The electronic band structure calculations were performed within 1500
points in the irreducible Brillouin zone (IBZ). The self-consistent calculations are converged since the total energy of the system is stable within 0.00001 Ry. The electronic band structure calculations were performed within 1500  points and the transport properties within 5000
points and the transport properties within 5000 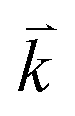 points in the IBZ.
points in the IBZ.
3. Results and discussion
3.1. Salient features of the electronic band structures
To ascertain the influence of compression mechanism on the transport properties we recall the calculated electronic band structure of CrF3 under different pressures. Since the area around the Fermi level (EF) plays an important role for carrier transportation, we concentrate our attention on this area. Fig. 1(a) and 1(b) illustrate the calculated electronic band structure in a wide energy range, and the enlarged area of the electronic band structure above and below EF. It is clear that the pressure causes a significant influence on the band location below and above the Fermi level (EF) resulting in some modifications to the ground state properties. It causes an increase in the parabolic shape of the bands in the vicinity of EF resulting in low effective mass and hence high mobility. The pressure leads to a reduction of the bond lengths and angles which results in increasing the orbital splitting, in agreement with the experimental data.3 As a result, the bonding states will go deeper into the lower energy level. Meanwhile, the energy level of the anti-bonding states will be enhanced. Also, the pressure pushes Cr-s/p orbitals to hybridize with the F-s orbital. This may lead to formation of partial covalent bonding between these orbitals which leads to changes in the chemical bonding character. Covalent bonding is more favorable for the transport of the carriers than ionic bonding.23 The bonds can significantly influence the carriers’ mobility and hence the electrical conductivity.23 A small octahedral strain was found when increasing the pressure which agrees well with the experimental data.3 From the calculated electronic band structures there are six bands crossing EF to form the shape of the Fermi surface. Therefore, the metallic nature of CrF3 corresponds to these six bands. In all cases the shape of the Fermi surface and the density of states at the Fermi level (N(EF)) originate mainly from Cr-d states with a small contribution of F-s/p and Cr-s/p states. | ||
| Fig. 1 Calculated electronic band structure for the pressure values 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8): (a) the calculated electronic band structure in a wide energy range; (b) the enlarged area of the electronic band structure above and below EF. | ||
3.2. Transport properties
 | (1) |
 | (2) |
In the above equations, the integral is performed over the Brillouin zone (BZ) as well as over the conduction band (CB) for electrons (e) or valence band (VB) for holes (h) and Ω is the volume of the unit cell. Dv(ε) and Dc(ε) are the density of states of the valence and the conduction bands. The charge carrier concentration in CrF3 as a function of temperature is illustrated in Fig. 2(a). It is clear that the charge carrier concentration decreases with increasing the temperature. Also it decreases with increasing pressure up to 1.84 GPa; then, after a notable increase in the carrier concentration occurs to reach the maximum value when the pressure rises up to 3.43 GPa, further increases in pressure cause a significant reduction in the carrier concentration to reach the lowest value at 5.43 GPa. Raising the pressure up to 8.56 GPa causes a huge increase in the carrier concentration. An increase in pressure up to 9.12 GPa leads to a drop in the carrier concentration to the lowest value which was already obtained at 5.43 GPa. Therefore, the pressure value 3.43 GPa is the optimal pressure to gain the highest carrier concentration. The values of the charge carrier concentration of CrF3 at 50 and 900 K under different pressure values are listed in Table 2. Following Fig. 2(a) and Table 2 we can conclude that the carrier concentration is irregularly changed by altering the pressure.
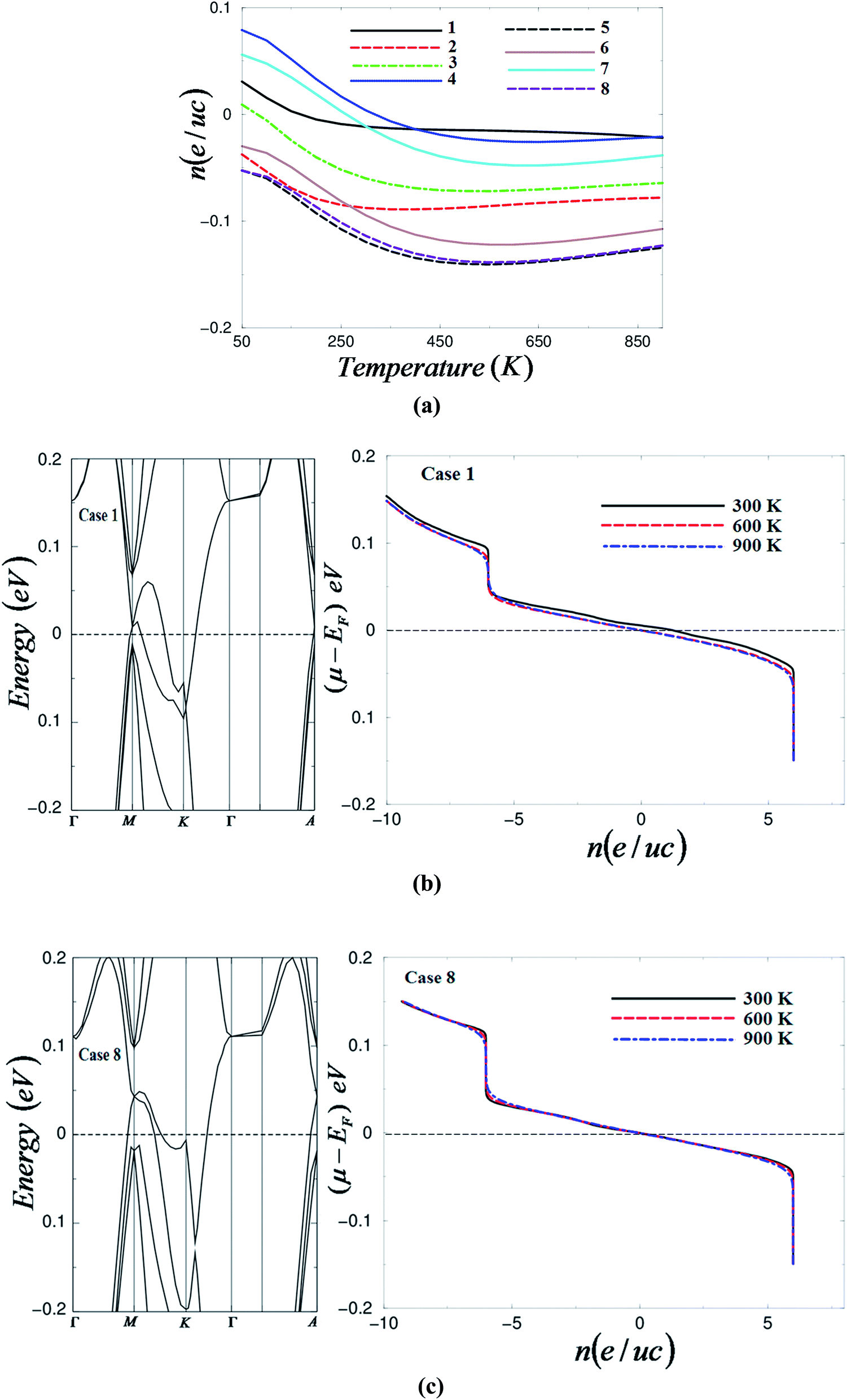 | ||
| Fig. 2 (a) Calculated carrier concentration as a function of temperatures under different pressure values; 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8); (b) band structure of CrF3 at ambient pressure together with the calculated number of carrier concentration at three constant temperatures (300, 600 and 900 K); (c) band structure of CrF3 at high pressure together with the calculated number of carrier concentration at three constant temperatures (300, 600 and 900 K). | ||
In Fig. 2(b) and (c) the electronic band structure of CrF3 in the energy region between 0.2 and −0.2 eV is plotted together with the carrier concentration at three constant temperatures (300, 600 and 900 K) at ambient and high pressure. The difference between the chemical potential and the Fermi energy (μ − EF) is positive for the valence band and negative for the conduction band. It is clear from the electronic band structure that CrF3 has parabolic bands around the Fermi level, therefore, the carriers exhibit low effective mass and hence high mobility. Both cases exhibit a maximum carrier concentration around the Fermi level and confirm that CrF3 possesses n-/p-type conduction in the chemical potential confined between ±0.2 eV, the region where CrF3 is expected to give maximum efficiency. At ambient pressure, changing the temperature causes a change in the carrier concentration (Fig. 2(b)) while at high pressure the temperature has no significant influence on the carrier concentration (Fig. 2(c)).
The electrical conductivity of CrF3 as a function of temperature under different pressure values at a certain chemical potential value (Table 2) is presented in Fig. 3(a). It has been found that a reduction in electrical conductivity occurs with an increase in temperature within the investigated pressure range, whereas the electrical conductivity significantly increases with an increase in pressure, which suggests that the CrF3 could be a good thermoelectric material within the investigated pressure range. The electrical conductivity values of CrF3 under different temperatures and pressures at a certain value of the chemical potential are listed in Table 2.
 | ||
| Fig. 3 (a) Calculated electrical conductivity as a function of temperatures under different pressure values; 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8); (b) calculated electrical conductivity as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under ambient pressure (case 1); (c) calculated electrical conductivity as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under a high pressure of 9.12 GPa (case 8). | ||
To ascertain the influence of compression mechanism on the electrical conductivity of CrF3, we have investigated the electrical conductivity as a function of chemical potential at three constant temperatures (300, 600 and 900 K). As an example we illustrated the influence of compression mechanism on the electrical conductivity at ambient and high pressure as shown in Fig. 3(b) and (c). It is clear that when increasing the pressure, a significant increase in the electrical conductivity occurs in the vicinity of EF that is attributed to the fact that the pressure causes an increase to the curvature of the parabolic shape of the bands in the vicinity of EF. Therefore, the carriers exhibit low effective mass and hence high mobility, which could be one of the important reasons for increasing the electrical conductivity according to the formula σ = neη. It has been noted that the highest value of σ/τ is achieved at 300 K for both ambient and high pressure which is about 1.538 × 1020 (Ω m s)−1 at μ − EF = −0.016 under ambient pressure and 1.418 × 1020 (Ω m s)−1 at μ − EF = −0.019 under high pressure, whereas it is about 2.675 × 1020 (Ω m s)−1 at μ − EF = +0.144 under ambient pressure and 1.969 × 1020 (Ω m s)−1 at μ − EF = +0.148 under high pressure.
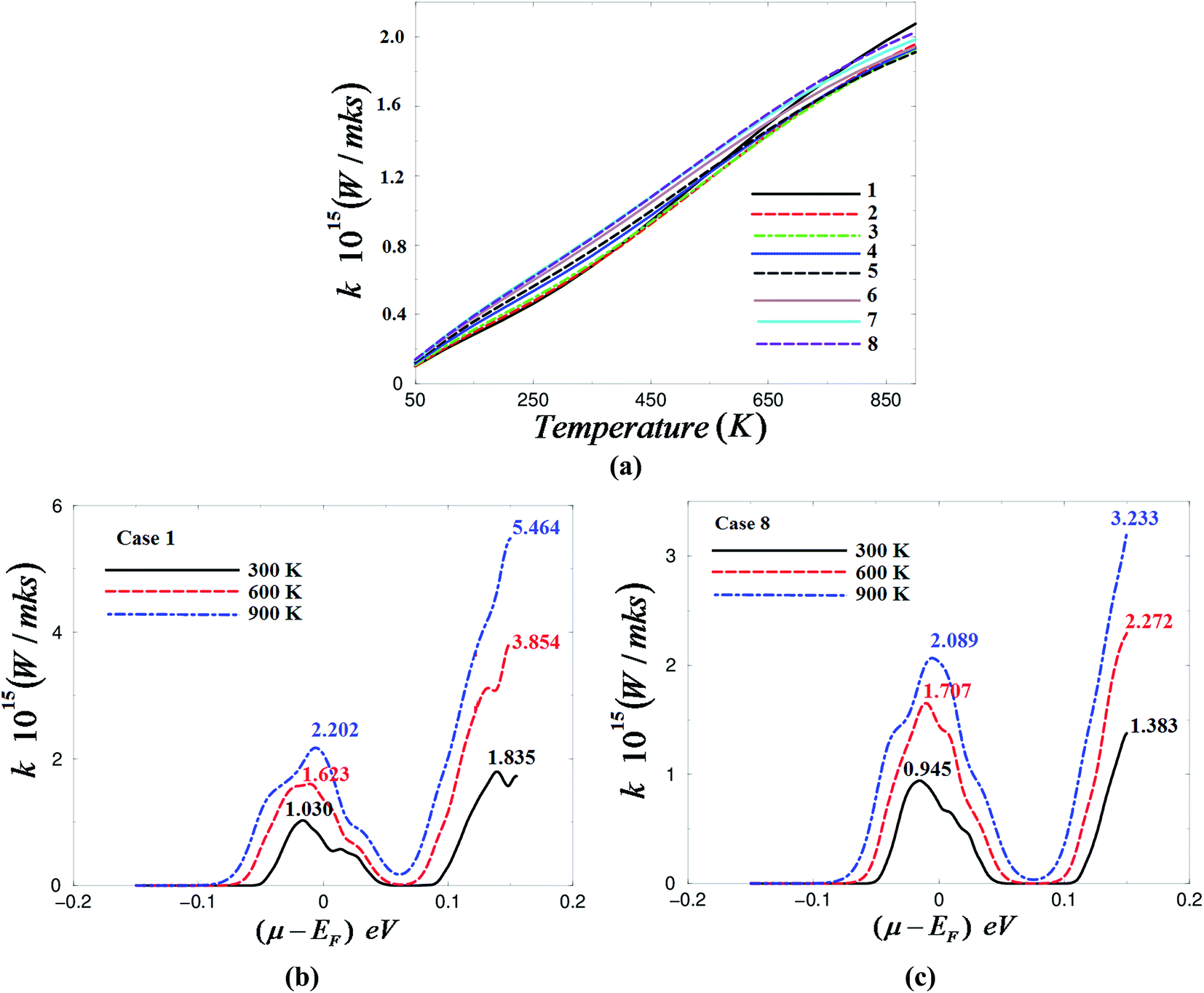 | ||
| Fig. 4 (a) Calculated electronic thermal conductivity as a function of temperatures under different pressure values; 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8); (b) calculated electronic thermal conductivity as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under ambient pressure (case 1); (c) calculated electronic thermal conductivity as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under a high pressure of 9.12 GPa (case 8). | ||
 | ||
| Fig. 5 (a) Calculated Seebeck coefficient as a function of temperatures under different pressure values; 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8); (b) calculated Seebeck coefficient as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under ambient pressure (case 1) along with the electronic band structure; (c) calculated Seebeck coefficient as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under a high pressure of 9.12 GPa (case 8) along with the electronic band structure. | ||
| P (GPa) | Seebeck coefficient (V K−1) @50 K | Seebeck coefficient (V K−1) @900 K | Power factor (mW m−1 K−2) @900 K |
|---|---|---|---|
| 0.0001 | −2.909 × 10−6 | 3.299 × 10−5 | 9.083 × 1010 |
| 1.01 | −1.559 × 10−7 | 3.087 × 10−5 | 8.071 × 1010 |
| 1.84 | −7.913 × 10−7 | 2.992 × 10−5 | 7.718 × 1010 |
| 3.43 | 1.114 × 10−6 | 2.875 × 10−5 | 7.482 × 1010 |
| 5.43 | −1.956 × 10−6 | 2.801 × 10−5 | 7.200 × 1010 |
| 7.46 | −1.744 × 10−6 | 2.727 × 10−5 | 7.200 × 1010 |
| 8.56 | −2.062 × 10−6 | 2.632 × 10−5 | 7.200 × 1010 |
| 9.12 | −1.532 × 10−6 | 2.621 × 10−5 | 7.059 × 1010 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 24,25 which shows that the power factor comes in the numerator of the figure of merit, thus the power factor is an important quantity and plays a principal role in evaluating the transport properties of the materials. The power factor of CrF3 is calculated at different temperatures and under different pressures for a fixed value of chemical potential, as shown in Fig. 6(a). It has been noted that the power factor is zero till 160 K, then above this temperature the power factor increases rapidly with increasing temperature. At high temperatures a reduction in power factor occurs with increasing pressure. The values of power factor at 900 K under different pressure values are listed in Table 3.
24,25 which shows that the power factor comes in the numerator of the figure of merit, thus the power factor is an important quantity and plays a principal role in evaluating the transport properties of the materials. The power factor of CrF3 is calculated at different temperatures and under different pressures for a fixed value of chemical potential, as shown in Fig. 6(a). It has been noted that the power factor is zero till 160 K, then above this temperature the power factor increases rapidly with increasing temperature. At high temperatures a reduction in power factor occurs with increasing pressure. The values of power factor at 900 K under different pressure values are listed in Table 3.
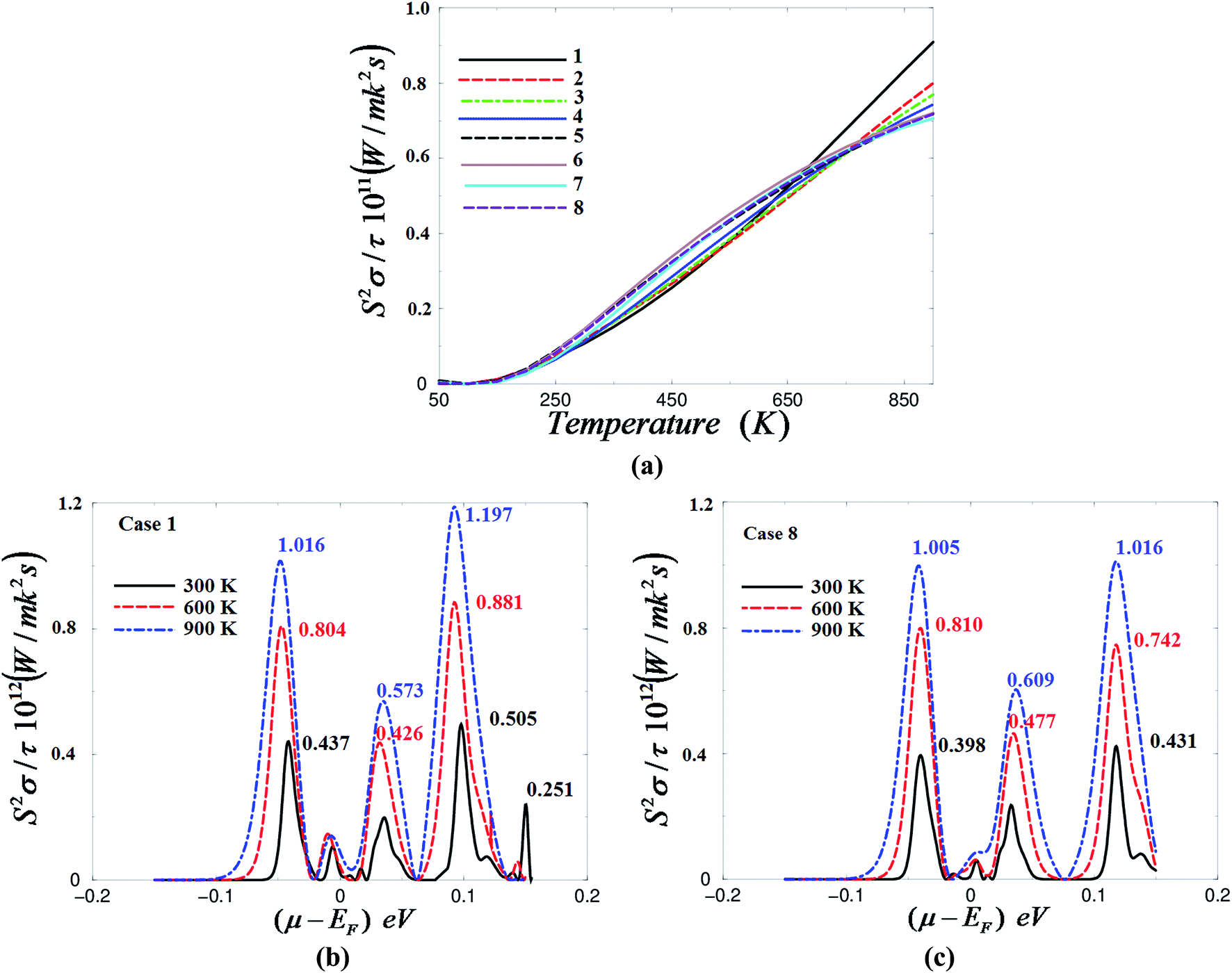 | ||
| Fig. 6 (a) Calculated power factor as a function of temperatures under different pressure values; 0.0001 GPa (case 1), 1.01 GPa (case 2), 1.84 GPa (case 3), 3.43 GPa (case 4), 5.43 GPa (case 5), 7.46 GPa (case 6), 8.56 GPa (case 7) and 9.12 GPa (case 8); (b) calculated power factor as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under ambient pressure (case 1); (c) calculated power factor as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under a high pressure of 9.12 GPa (case 8). | ||
In order to ascertain the influence of the compression mechanism on the power factor of CrF3, we have calculated the power factor at 300, 600 and 900 K as a function of chemical potential under different pressures, as presented in Fig. 6(b) and (c). It is clear that the pressure has a significant influence on the power factor and causes the structures around μ − EF = +0.1 eV to shift towards a higher chemical potential. The power factor values are mentioned directly on each peak of the power factor in Fig. 6(b) and (c).
4. Conclusions
The all-electron full potential linear augmented plane wave (FPLAPW+lo) method in a scalar relativistic version as embodied in the WIEN2k code is used to obtain the electronic structure of CrF3 at different pressures. The electronic transport coefficients were evaluated by utilizing the semi-classical Boltzmann theory and rigid band model. The carrier concentration (n), Seebeck coefficient (S), electrical conductivity (σ/τ), electronic thermal conductivity (κe/τ), and the electronic power factor (S2σ/τ) as a function of temperature at a certain value of chemical potential as well as a function of chemical potential at three constant temperatures (300, 600 and 900 K) under different pressure values are theoretically investigated. It has been noted that the pressure value 3.43 GPa is the optimal value to gain the highest carrier concentration. A reduction in electrical conductivity occurs with increasing temperature within the investigated pressure range and the electrical conductivity significantly increases with increasing pressure. The Seebeck coefficient represents n-/p-type conductions. The electronic thermal conductivity ke increases linearly with increasing temperature under different pressure values. The power factor is zero till 160 K, then increases rapidly with increasing temperature. At high temperatures a reduction in the power factor occurs with increasing pressure.Acknowledgements
The results were developed within the CENTEM project, reg. no. CZ.1.05/2.1.00/03.0088, co-funded by the ERDF as part of the Ministry of Education, Youth and Sports OP RDI programme and, in the follow-up sustainability stage, supported through CENTEM PLUS (LO1402) by financial means from the Ministry of Education, Youth and Sports under the ”National Sustainability Programme I. Computational resources were provided by MetaCentrum (LM2010005) and CERIT-SC (CZ.1.05/3.2.00/08.0144) infrastructures.References
- K. H. Jack and R. Maitland, Proc. Chem. Soc., London, 1957, 232 CAS.
- K. Knox, Acta Crystallogr., 1960, 13, 507–508 CrossRef CAS.
- J.-E. Jorgensen, W. G. Marshall and R. I. Smith, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 669–673 CAS.
- W. Fischer, Z. Kristallogr., 1973, 138, 129–146 CrossRef CAS.
- K. Meisel, Z. Anorg. Allg. Chem., 1932, 207, 121–128 CrossRef CAS PubMed.
- P. Herzig and J. Zemann, Z. Kristallogr., 1993, 205, 85–97 CrossRef CAS PubMed.
- R. von Losch, C. Hebecher and Z. Ranft, Z. Anorg. Allg. Chem., 1982, 491, 199–202 CrossRef PubMed.
- A. A. Hepworth, K. H. Jack, R. D. Peacock and G. J. Westland, Acta Crystallogr., 1957, 10, 63–69 CrossRef.
- L. Grosse and R. Hoppe, Z. Anorg. Allg. Chem., 1987, 552, 123–131 CrossRef CAS PubMed.
- H. Sowa and H. Ahsbahs, Acta Crystallogr., Sect. B: Struct. Sci., 1998, 54, 578–584 CrossRef.
- J.-E. Jorgensen, J. S. Olsen and L. Gerward, J. Appl. Crystallogr., 2000, 33, 279–284 CrossRef CAS.
- G. K. H. Madsen and D. J. Singh, Comput. Phys. Commun., 2006, 175, 67–71 CrossRef CAS PubMed.
- A. H. Reshak and S. Auluck, RSC Adv., 2014, 4, 37411 RSC.
- A. H. Reshak, RSC Adv., 2014, 4, 39565 RSC.
- M. Jamal, N. Kamali Sarvestani, A. Yazdani and A. H. Reshak, RSC Adv., 2014, 4, 57903 RSC.
- A. H. Reshak, RSC Adv., 2014, 4, 63137 RSC.
- B. Xu, X. Li, G. Yu, J. Zhang, S. Ma, Y. Wang and L. Yi, J. Alloys Compd., 2013, 565, 22–28 CrossRef CAS PubMed.
- D. Wang, L. Tang, M. Q. Long and Z. G. Shuai, J. Chem. Phys., 2009, 131, 224704 CrossRef PubMed.
- T. J. Scheidemantel, C. Ambrosch-Draxl, T. Thonhauser, J. V. Badding and J. O. Sofo, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 125210 CrossRef.
- G. J. Snyder and E. S. Toberer, Nat. Mater., 2008, 7, 105–114 CrossRef CAS PubMed.
- P. Blaha, K. Schwarz, G. K. H. Madsen, D. Kvasnicka and J. Luitz, WIEN2k, An augmented plane wave plus local orbitals program for calculating crystal properties, Vienna University of Technology, Austria, 2001 Search PubMed.
- J. P. Perdew, S. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- F. Wu, H. z. Song, J. f. Jia and X. Hu, Prog. Nat. Sci., 2013, 23(4), 408–412 CrossRef PubMed.
- C. C. Hu, Modern Semiconductor Devices for Integrated Circuits, Part I: Electrons and holes in a semiconductor, November 11, 2011 Search PubMed.
- J.-H. Lee, J. Wu and J. C. Grossman, Phys. Rev. Lett., 2010, 104, 016602 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |