
Li4Ti5O12 hollow mesoporous microspheres assembled from nanoparticles for high rate lithium-ion battery anodes†
Liyun Cao,
Yani Hui,
Haibo Ouyang,
Jianfeng Huang*,
Zhanwei Xu,
Jiayin Li,
Wanzhuo Zhang,
Simin Chai and
Shouwu Guo
School of Materials Science & Engineering, Shaanxi University of Science and Technology, Xi'an 710021, China. E-mail: huangjfsust@126.com; Fax: +86 029 86168802; Tel: +86 029 86168802
First published on 14th April 2015
Abstract
Li4Ti5O12 hollow mesoporous microspheres (HMMs) assembled from nanoparticles were successfully synthesized by a facile hydrothermal method and subsequent calcination. They exhibit superior rate capabilities with reversible capacities of 176, 125 and 86 mA h g−1 after 10 cycles at 0.1 C, 20 C and 40 C (7000 mA g−1), respectively. The Li4Ti5O12 HMMs also possess outstanding cycle performance with only 3% capacity degradation at 10 C after 500 cycles, which is equivalent to a fade of merely 0.006% per cycle. The comparison with other studies shows that the Li4Ti5O12 HMMs possess more promising reversible capacity and rate capability among the values reported for Li4Ti5O12. These excellent electrochemical properties may be attributed to the unique HMM structures.
Introduction
Lithium-ion batteries (LIBs) have become excellent power sources for portable electronic devices and hybrid electric vehicles due to their relatively high energy/power density, low self-discharge and the absence of memory effects.1–5 The carbon-based material has been commonly adopted as an anode material in most commercial LIBs, since it shows a desirable charge potential profile and long cycling life.6,7 However, carbonaceous anodes generally suffer from poor coulombic efficiencies at high C-rates and serious safety problems originated from excessive lithium dendrite formation on the anode surface after long-term charge–discharge operations.8–11 Therefore, there is an urgency to develop alternative anode materials with improved rate and safety performances. A variety of nanomaterials, such as Co3O4,12,13 ZnO2,14,15 SnO2,16–18 TiO2,19,20 SiO2 (ref. 21 and 22) and VO2 (ref. 23 and 24) have been intensively investigated as anode materials for LIBs. Nevertheless, most of these materials usually possess poor cycling performance caused by large volume expansion – shrinkage during Li-ion intercalation–extraction reactions, which predisposes the material to pulverization and loss of electrical connectivity.25,26As a promising substitute for graphite, spinel-structured Li4Ti5O12 has attracted much interest because of several attractive advantages.27–31 It possesses an extremely flat voltage plateau at about 1.55 V (vs. Li+/Li), which suppresses the decomposition of electrolyte and the deposition of lithium dendrites, making the LIBs safe.32,33 Furthermore, Li4Ti5O12 also displays excellent cycling stability and outstanding structural stability due to the zero volume change during Li+ insertion/extraction.25,34 However, its low electronic conductivity (<10−13 S cm−1) and Li+ diffusion coefficient (10−9 to 10−13 cm2 s−1) result in poor rate performance,35,36 which limits its practical applications in the fields of electric vehicles and hybrid electric vehicles.
In recent years, extensive efforts have been devoted to improve the rate capability of Li4Ti5O12, and one of the most common strategies is to fabricate nanostructured Li4Ti5O12.37–39 Since the diffusion capability of the ion is in inverse proportion to square of particle size and in proportion to diffusion coefficient,40 the decrease in the grain size can significantly enhance the rate performance of Li4Ti5O12. Up to now, the mesoporous structure assembled by nanounits has been regarded as an ideal host for the rapid transportation of both ions and electrons in high-rate LIBs,41–43 as it possesses both the advantages of nanometer-sized building blocks (e.g., shortened diffusion distance and high specific area) and micrometer-sized assemblies (e.g., excellent thermodynamic stability and high tap-density), benefiting for cycling and rate performances.44 Moreover, its porosity structures would allow electrolyte to penetrate easily, which makes the Li+ insertion/extraction more sufficiently.45
In order to further optimize the structures and electrochemical performances of mesoporous microsphere, such as decrease its density and make the best of its central nanounits, more and more researchers turned their attention to the preparation of hollow mesoporous microspheres. Li4Ti5O12 hollow microspheres assembled by nanoparticles46 or nanosheets47 have been successfully prepared, exhibiting superior high rate capability and great cycling stability. However, templates or high reaction temperatures are usually needed in the system, which makes the synthetic process complicated and costly. Therefore, in this work, we employed a facile hydrothermal method and following calcinations yielding unique Li4Ti5O12 hollow mesoporous microspheres (HMMs) assembled by nanoparticles without templates or surfactants. The obtained Li4Ti5O12 HMMs, used as anode materials in LIBs, exhibited superior cycle and rate performances.
Experimental
Material preparation
Li4Ti5O12 HMMs were synthesized by a hydrothermal process and following calcinations. The lithium hydroxide monohydrate (LiOH·H2O) was purchased from China National Medicines Corp., Ltd. The precursor TiO2 was prepared using the method in ref. 48. Typical, 0.5 g TiO2 was added into a 40 ml 2 M LiOH solution under magnetic stirring for 30 min. The mixture was transferred into a 100 ml Teflon-lined autoclave in a homogeneous reactor at 160 °C for 48 hours. After cooling down naturally, the resulting precipitate was collected by centrifugation and washed thoroughly with deionized water for several times. Eventually, the obtained products after hydrothermal process was calcined at 400 °C (200 °C, 600 °C and 900 °C are shown in ESI†) for 2 h.Materials characterization
Crystal structures of the prepared samples were characterized by X-ray diffraction (XRD) using an X-ray powder diffractometer (Rigaku D/max-2200PC). X-ray profiles were recorded between 15° and 70° (2θ) with Cu Kα radiation (λ = 1.5406 Å). The Raman spectra were recorded with a confocal microprobe Raman system (Renishaw-invia with a laser at 532 nm). Particle sizes, morphologies, and microstructures of the samples were observed by using a field-emission scanning electron microscope (FESEM, S-4800) and a transmission electron microscope (TEM, TecnaiG2F20S-TWIN). Nitrogen adsorption/desorption isotherms were acquired at 77 K using a surface area and pore size analyser (NOVA 2200e). Specific surface areas were calculated based on the Brunauer–Emmett–Teller (BET) method. The Barrett–Joyner–Halenda (BJH) pore size distribution was determined from the desorption isotherms. Thermal gravimetric-differential scanning calorimetry analysis (TGA-DSC) was performed on a STA409PC, in nitrogen with a heating rate of 10 °C min−1.Electrochemical measurements
Electrochemical measurements were performed using CR2032 coin-type cells assembled in an argon-filled glove box. To prepare anode materials as working electrode, the synthesized Li4Ti5O12, polyvinylidene fluoride (PVDF) binder and acetylene black were mixed together at a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 in N-methylpyrrolidinone (NMP). The slurry was then coated onto copper foil. After vacuum drying at 80 °C for 24 h, electrode discs with a diameter of 16 mm (area of about 2 cm2) were punched out and weighed. The mass of Li4Ti5O12 material was around 3.0 mg. The electrolyte consisted of a solution of 1 M LiPF6 in ethylene carbonate and dimethyl carbonate (EC + DMC, 1:1 in volume). The electrochemical performance of the Li4Ti5O12 HMM was examined with lithium as counter electrode. The capacity and cycle performance were evaluated by a multichannel battery testing system (Shenzhen, Neware, China) with a potential window ranging from 1.0 to 2.5 V at various current densities. Cyclic voltammograms (CV) were recorded at scan rates of 0.05 mV s−1, using CHI660E electrochemical station (Shanghai Chenhua, China). All above electrochemical measurements were taken at room temperature.
:10:10 in N-methylpyrrolidinone (NMP). The slurry was then coated onto copper foil. After vacuum drying at 80 °C for 24 h, electrode discs with a diameter of 16 mm (area of about 2 cm2) were punched out and weighed. The mass of Li4Ti5O12 material was around 3.0 mg. The electrolyte consisted of a solution of 1 M LiPF6 in ethylene carbonate and dimethyl carbonate (EC + DMC, 1:1 in volume). The electrochemical performance of the Li4Ti5O12 HMM was examined with lithium as counter electrode. The capacity and cycle performance were evaluated by a multichannel battery testing system (Shenzhen, Neware, China) with a potential window ranging from 1.0 to 2.5 V at various current densities. Cyclic voltammograms (CV) were recorded at scan rates of 0.05 mV s−1, using CHI660E electrochemical station (Shanghai Chenhua, China). All above electrochemical measurements were taken at room temperature.
Results and discussion
Structure and morphology
X-ray diffraction (XRD) analysis was employed to investigate the precursor phases. The XRD patterns of the precursor TiO2 is shown in Fig. 1a. It is found that the titanium source in this experiment is pure rutile TiO2 and the wide diffraction peaks may imply the grain size is very small. The morphology and microstructure of the precursor was examined by SEM and TEM. Fig. 1b shows a SEM image of the TiO2, which is composed of microspheres with a rough surface. Many of the microspheres are closely connected, and the diameter of the microsphere is around 1.3 μm. Fig. 1c and d reveal that the TiO2 microsphere is actually is comprised of the assembly of interconnected nanorods with a diameter of about 20 nm. The nanocrystallite sizes are in good agreement with that estimated from XRD. | ||
| Fig. 1 XRD patterns (a), SEM images (b) and (c), TEM images (d) of the precursor TiO2 (inset: magnified TEM image of the second red rectangle). | ||
The XRD patterns of the products after hydrothermal process (PAHP) and the Li4Ti5O12 calcined at temperatures of 300 °C, 600 °C and 900 °C are shown in Fig. S1 (ESI†), while the Li4Ti5O12 calcined at 400 °C is shown in Fig. 2a. It is found that the calcination temperature greatly affected the crystal phases of the final products. As shown in Fig. S1,† the diffraction peaks of the PAHP conform to a LiTiO2 (JCPDS no. 16-0223) without obvious impurity phase. After calcination at 300 °C, the final products exhibit a weak and broadened peak at approximately 18.3°, which can be indexed to spinel Li1+xTi2−yO4+δ.49 When the calcination temperature rises to 400 °C, all the diffraction peaks of the final products correspond well with a face-centered cubic spinel structure of Li4Ti5O12 (JCPDS no. 49-0207), which indicates that the PAHP is completely converted into the crystallized spinel Li4Ti5O12. Eventually, the Li4Ti5O12 is partly decomposed into LiTiO2 and TiO2 after calcined at 900 °C.
 | ||
| Fig. 2 XRD patterns (a), Raman spectrum (b), SEM images (c) and (d), TEM images (e), HRTEM images (f), SAED patterns (g), nitrogen adsorption/desorption isotherms, small-angle XRD patterns (inset) (h) and pore size distribution (i) of the Li4Ti5O12. | ||
Moreover, the TG-DSC test of the products after hydrothermal process is shown in Fig. S2 (ESI†). The TG curve in Fig. S2† shows a weight loss of around 13% from room temperature to 300 °C. As can be seen in the DSC curve, there is an obvious endothermal peak at 200–400 °C and an exothermal peak at approximately 900 °C in the DSC curves. Correspond to the TG curve, the flat plateau around 400 °C indicates the formation of Li4Ti5O12 which is confirmed in the XRD pattern of the 400 °C product. The beginning of the Li4Ti5O12 decomposition locates at the range of 600–800 °C.
The structure of Li4Ti5O12 calcined at 400 °C is further confirmed by the results of Raman spectrum plotted in Fig. 2b. The products exhibit five vibration peaks at 230.8, 276.5, 335.8, 429.9 and 686.3 cm−1, which can be indexed to the A1g + Eg + 3F2g spinel structure. The peak at 686.3 cm−1 is characteristic of A1g mode, which can be assigned to Ti–O stretches in “TiO6” octahedral. The peak at 429.9 cm−1 is ascribed to Eg mode, and the other three peaks are F2g modes.28 In addition, no obvious peaks were detected, confirming the high purity phase in the products. It was demonstrated that spinel Li4Ti5O12 without any distinct impurities could be obtained after the calcinations at 400 °C greatly lower than the solid state synthesis.7,8,33,34,45–47,49,50
To further examine the architecture of the Li4Ti5O12, the samples were investigated by SEM, TEM and HRTEM. The SEM images of the PAHP and the Li4Ti5O12 calcined at temperatures of 300 °C, 600 °C and 900 °C are shown in Fig. S3 (ESI†), while the Li4Ti5O12 calcined at 400 °C is shown in Fig. 2c and d. It shows that the morphologies of PAHP turn out to be hollow microspheres, the shells of which are assembled by many nanoparticles. Similar hollow microsphere structure can be obtained after calcined at 300 °C, 400 °C and 600 °C, except for 900 °C, as shown in Fig. 2c and S3.† Fig. 2c shows that the Li4Ti5O12 HMMs are about 1.2 μm in diameter, with a shell thickness of approximately 400 nm. Upon closer examination the shell was found to be assembled by thousands of nanoparticles with the size of around 60 nm (Fig. 2d). Furthermore, many of the microspheres are broken and the cavities can be clearly seen from Fig. 2c. Further evidence for the hollow structure can also be found from Fig. 2e. The TEM image of the Li4Ti5O12 HMMs is shown in Fig. 2e. The clear contrast between the dark edge and the gray center of each microsphere reveals its hollow nature; the gray parts and white spots in the center of every microsphere confirms the presence of porous structure. The HRTEM image in Fig. 2f shows that the interplanar distance between adjacent lattice fringes is 0.48 nm, corresponding to the (111) interplanar spacing of spinel Li4Ti5O12, which indicates the Li4Ti5O12 HMMs are assembled by the well-crystallized spinel Li4Ti5O12 nanoparticles.50 The clear contrast of light and shade between the primary particles can be found in the HRTEM image shown in Fig. S4 (ESI†), indicating the presence of three-dimensionally interconnected mesopores in the Li4Ti5O12 HMMs. The formation mechanism of the hollow structured Li4Ti5O12 was presented in ESI.† The corresponding selected area electron diffraction (SAED) pattern is demonstrated in Fig. 2g, which can be indexed to the diffraction planes of (111), (222), (331) and (531) of the spinel Li4Ti5O12 phase, suggesting the polycrystalline nature of the Li4Ti5O12 HMMs.
N2 adsorption/desorption analysis was used to examine the mesoporous structure of Li4Ti5O12, as shown in Fig. 2h. The isotherm curve seemed to be the intermediate between type II and IV51 with an H3 hysteresis loop, which exhibited the hysteresis loop beginning at relative pressures of P/P0 = 0.44, but without dramatic flat desorption isotherms at high relative pressures of P/P0 = 0.9–1.0. The small-angle XRD patterns of Li4Ti5O12 is illustrated in the inset of Fig. 2h. It is seen that the Li4Ti5O12 presented one intense diffraction peak, indicating that a well-organized mesoporous structure was formed,44 which was also confirmed by TEM micrographs (Fig. 2e) and HRTEM micrographs (Fig. S4†). The Barrett–Joyner–Halenda (BJH) pore size distribution of the Li4Ti5O12, shown in Fig. 2i, indicated that the mesoporous Li4Ti5O12 materials exhibited a broad range of pore sizes (2.9–62.1 nm) distribution, with the average pore diameter of 3.8 nm. According to Brunauer–Emmett–Teller (BET) analysis, a large specific surface area of 86.5 m2 g−1 is obtained, benefiting its lithium storage.52,53
Electrochemical performance
To test the potential application of the Li4Ti5O12 HMMs in Li ion batteries, we investigated their electrochemical performance toward Li insertion/extraction. Fig. 3a shows the first three consecutive cyclic voltammograms of the Li4Ti5O12 HMM electrodes at a scan rate of 0.05 mV s−1 in the potential range from 2.5 to 1.0 V (vs. Li+/Li). The reduction/oxidation peaks around 1.55 V can be observed during the charge/discharge process, which indicates the characteristic of the two-phase reaction between Li7Ti5O12 and Li4Ti5O12.52 Meanwhile, the three CV curves are over-lapped, suggesting excellent cycle stability.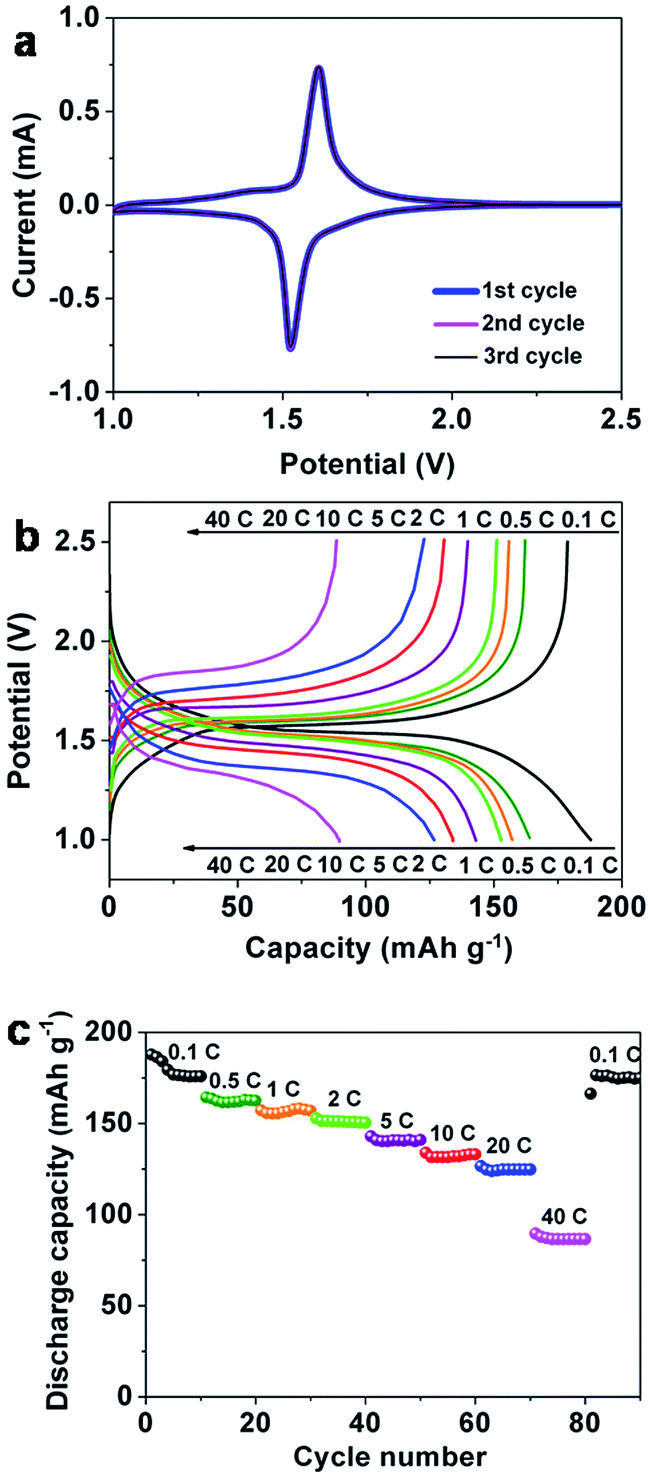 | ||
| Fig. 3 The first three consecutive cyclic voltammograms of the Li4Ti5O12 (a); initial charge/discharge curves of the Li4Ti5O12 at 0.1–40 C (b); rate properties of the Li4Ti5O12 at 0.1–40 C (c). | ||
Fig. 3b presents the charge/discharge profiles of the Li4Ti5O12 HMM electrodes cycled at current rates from 0.1 to 40 C. The charge/discharge voltage profiles show a pair of voltage plateaus at the potential around 1.55 V, which is consistent with the CV results. The initial discharge and charge capacities at 0.1 C are 188 and 179 mA h g−1, corresponding to the initial coulombic efficiency (charging capacity/discharging capacity) of 95%, implying little initial irreversible capacity loss. The high initial coulombic efficiency and charging/discharging capacity indicate that lithium ions could almost totally extract after lithium insertion and the active sites would be nearly fully utilized. With increasing of the charge/discharge rates, the potential differences between charge and discharge plateaus exhibit a tendency to increase which could be attributed to the increased electrode polarization and the sluggish diffusion kinetics of Li-ion at high rates.6
The corresponding rate performance is shown in Fig. 3c. It was tested at 0.1 C for the first 10 cycles, and then, the rate was increased to 40 C. At the rate of 0.1 C, the discharge capacity is gradually decrease from 188 to 176 mA h g−1 within the first 4 cycles. However the discharge capacities remain stable maintaining a value of 176 mA h g−1 for the next 6 cycles. The discharge capacity of the Li4Ti5O12 HMM electrodes at 0.1 C is slightly higher than its theoretical value (175.4 mA h g−1); many researchers have reported this phenomenon as well.28,33,38 It may be due to the electrode material's unique morphology, large surface area, lattice defect, or impurity phases within the electrode materials. The discharge capacity of 163 mA h g−1 is obtained at a rate of 0.5 C after 10 cycles; the capacities are 157, 151, 141, 133, 125 and 86 mA h g−1 when tested at 1, 2, 5, 10, 20 and 40 C, respectively. It is worth noting that the capacity can be completely recovered to 174 mA h g−1 when the discharge rate returns to 0.1 C rate after continuous 40 C rate cycles, indicating its good electrochemical reversibility even after high rate discharge–charge cycles. The comparison of rate properties between the three samples annealed at 300 (LTO – 300), 400 (LTO – 400, Li4Ti5O12 HMM electrodes) and 600 °C (LTO – 600) is shown in Fig. S6 (ESI†). It shows that the LTO – 400 exhibited significantly better rate performance than LTO – 300 and LTO – 600; the discharge capacities were in the following descending order: LTO – 400 > LTO – 600 > LTO – 300.
Cycling performance of the Li4Ti5O12 HMM electrodes at 10 C, 20 C and 40 C is illustrated in Fig. 4. The corresponding coulombic efficiency is shown in Fig. S4.† At the rate of 10 C, the discharge capacity gradually increases from 132 to 134 mA h g−1 between the 1st and 10th cycles and decreases from 134 to 130 mA h g−1 after the next 490 continuous cycles with only 3% degradation, which is equivalent to a capacity fade of merely 0.006% per cycle. They also exhibit reversible capacities 115 and 83 mA h g−1 after 500 cycles at 20 C and 40 C, respectively. As can be seen that capacity of the Li4Ti5O12 HMM electrodes remain stable, when the C rate increases, which may be due to that only part of the electrode material with the most electrochemical active actually charge/discharge in the test since Li+ ions and electrons don't have abundant time to diffuse and to conduct in the particles at higher C rate.2 The coulombic efficiencies at 10 C, 20 C and 40 C all stabilize above 99% between the 1st and 500th cycles, as shown in Fig. S5 (ESI†). These results demonstrate excellent cycling performance of the Li4Ti5O12 HMM electrodes even at the rate of 40 C.
 | ||
| Fig. 4 Cycling performance for the 500 cycles at the rates of 10 C, 20 C and 40 C, respectively. | ||
Fig. 5 and Table 1 compare the discharging capacity performance of Li4Ti5O12 HMM electrodes with recently reported Li4Ti5O12 electrodes.28,45,49,50,53–57 In order to make the table not too large, Table 1 only shows a part of the discharging capacity values. Fig. 5 graphically contrasts the discharging capacity versus C rates, indicating promising rate characteristics of the Li4Ti5O12 HMM electrodes.
 | ||
| Fig. 5 Discharging capacity performance comparison of Li4Ti5O12 HMM electrodes with recently reported Li4Ti5O12 electrodes, all tested as half cells vs. Li/Li+. | ||
| Material content | Synthesis method | Microstructure | Capacitya (mA h g−1) | C rate | Current collector | References |
|---|---|---|---|---|---|---|
| a The capacity after (X) cycles. | ||||||
| Pure Li4Ti5O12 | Hydrothermal method and following calcinations | HMMs assembled by nanoparticles | 157 (10) | 1 C | Copper foil | This work |
| 133 (10) | 10 C | |||||
| 125 (10) | 20 C | |||||
| 86 (10) | 40 C | |||||
| Pure Li4Ti5O12 | Microwave-assisted hydrothermal and microwave post annealing process | Microspheres composed of nanoflakes | 121 (10) | 10 C | Copper foil | Chou et al.28 |
| 98 (10) | 20 C | |||||
| 62 (10) | 40 C | |||||
| Li4Ti5O12 with tiny amounts of TiO2 | Molten salt process | Hierarchical mesoporous microspheres | 123 (10) | 8 C | Copper foil | Nugroho et al.45 |
| 103 (10) | 10 C | |||||
| 80 (10) | 20 C | |||||
| Li4Ti5O12 with little amounts of TiO2 | Solvothermal method and following calcinations | Mesoporous microspheres | 169 (initial) | 2 C | Aluminium plate | Lin et al.49 |
| 115 (initial) | 10 C | |||||
| Li4Ti5O12 with small amounts of Li2TiO3 | Hydrothermal synthesis and following calcinations | Hierarchically porous microspheres | 166 (10) | 0.2 C | Aluminium foil | Shen et al.50 |
| 144 (10) | 3 C | |||||
| 92 (10) | 20 C | |||||
| Pure Li4Ti5O12 | Hydrothermal synthesis and following calcinations | Mesoporous | 120 (20) | 3 C | Never mentioned | Lin et al.54 |
| 105 (20) | 10 C | |||||
| 83 (20) | 20 C | |||||
| 80 (20) | 30 C | |||||
| Li4Ti5O12 with little amounts of Li2CO3 | Coprecipitation method and following calcinations | Mesoporous nanoclusters | 159 (5) | 1 C | Copper foil | Sun et al.53 |
| 140 (5) | 2 C | |||||
| 138 (5) | 5 C | |||||
| Pure Li4Ti5O12 | High-energy ball milling process | Microsized particle | 133 (5) | 3 C | Aluminium foil | Liu et al.55 |
| 121 (5) | 5 C | |||||
| 93 (5) | 10 C | |||||
| Pure Li4Ti5O12 | Spray drying process | Nanosized particle | 174 (initial) | 0.1 C | Copper foil | He et al.56 |
| 136 (initial) | 5 C | |||||
| Pure Li4Ti5O12 | Sol–gel strategy | Monodispersed mesoporous | 160 (20) | 0.5 C | Never mentioned | Lin et al.57 |
| 107 (20) | 3 C | |||||
| 93 (20) | 5 C | |||||
It is well known that the morphology and particle size have important effects on the electrochemical properties of Li4Ti5O12. The HMM structure of Li4Ti5O12 have several merits to account for the excellent lithium storage properties and the much improved rate performances. Firstly, the open HMM structure (about 400 nm in diameter) would allow lithium (around 0.1 nm in diameter58) to insert into the Li4Ti5O12 particles from both inside and outside, which makes it more effective for lithium intercalation. Secondly, the large amount of pores, with average pore diameter of 3.8 nm, is desirable to accommodate a great number of Li ions within the pore not only by surface adsorption. This thesis has been proved in ref. 59. Thirdly, the interconnected nanoparticles could increase electrode/electrolyte contact areas and short diffusion paths for both Li+ and electrons within particles. The interconnected nanoparticles also significantly decrease their contact resistance, this view has been confirmed in ref. 60, thereby enhancing the high rate performance of Li4Ti5O12 HMM electrodes.
Conclusions
In summary, we obtained Li4Ti5O12 HMMs by a hydrothermal method and following calcinations. The Li4Ti5O12 HMMs are about 1.2 μm in diameter, and their shells are assembled by thousands of nanoparticles with the size of around 60 nm. Moreover, they deliver a reversible capacity as high as 176 mA h g−1 at the C rate of 0.1 C after 10 cycles. The Li4Ti5O12 HMM electrodes also exhibit reversible capacities 130, 115 and 83 mA h g−1 after 500 cycles at 10 C, 20 C and 40 C, respectively. A comparison with recently reported Li4Ti5O12 electrodes reveals that these performance matrices are highly favorable, indicating their promising applications for high power LIBs in the future.Acknowledgements
This work was supported by National Key Technology R&D Program (2013BAF09B02), National Natural Science Foundation of China (51472152), 973 Special Preliminary Study Plan (2014CB260411), Innovation Team Assistance Foundation of Shaanxi Province (2013KCT-06) and Graduate Innovation Fund of Shaanxi University of Science and Technology.References
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- C. Lin, X. Fan, Y. Xin, F. Cheng, M. O. Lai, H. Zhou and L. Lu, J. Mater. Chem. A, 2014, 2, 9982 CAS.
- M. Chen, W. Li, X. Shen and G. Diao, ACS Appl. Mater. Interfaces, 2014, 6, 4514 CAS.
- J. Haruyama, K. Sodeyama, L. Han, K. Takada and Y. Tateyama, Chem. Mater., 2014, 26, 4248 CrossRef CAS.
- Q. Yue, H. Jiang, Y. Hu, G. Jia and C. Li, Chem. Commun., 2014, 50, 13362 RSC.
- T. F. Yi, Z. K. Fang, Y. Xie, Y. R. Zhu and S. Y. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 20205 CAS.
- X. Li, H. C. Lin, W. J. Cui, Q. Xiao and J. B. Zhao, ACS Appl. Mater. Interfaces, 2014, 6, 7895 CAS.
- S. Chen, Y. Xin, Y. Zhou, Y. Ma, H. Zhou and L. Qi, Energy Environ. Sci., 2014, 7, 1924 CAS.
- J. Hassoun, F. Bonaccorso, M. Agostini, M. Angelucci, M. G. Betti, R. Cingolani, M. Gemmi, C. Mariani, S. Panero, V. Pellegrini and B. Scrosati, Nano Lett., 2014, 14, 4901 CrossRef CAS PubMed.
- Z. Jian, M. Zheng, Y. Liang, X. Zhang, S. Gheytani, Y. Lan, Y. Shi and Y. Yao, Chem. Commun., 2015, 51, 229 RSC.
- J. W. Lee, S. Y. Lim, H. M. Jeong, T. H. Hwang, J. K. Kang and J. W. Choi, Energy Environ. Sci., 2012, 5, 9889 CAS.
- W. H. Ryu, T. H. Yoon, S. H. Song, S. Jeon, Y. J. Park and I. D. Kim, Nano Lett., 2013, 13, 4190 CrossRef CAS PubMed.
- J. Jiang, Y. Li, J. Liu, X. Huang, C. Yuan and X. W. Lou, Adv. Mater., 2012, 24, 5166 CrossRef CAS PubMed.
- D. Bresser, F. Mueller, M. Fiedler, S. Krueger, R. Kloepsch, D. Baither, M. Winter, E. Paillard and S. Passerini, Chem. Mater., 2013, 25, 4977 CrossRef CAS.
- Y. Feng, R. Zou, D. Xia, L. Liu and X. Wang, J. Mater. Chem. A, 2013, 1, 9654 CAS.
- J. H. Kang, S. M. Paek and J. H. Choy, Chem. Commun., 2012, 48, 458 RSC.
- L. Wang, D. Wang, Z. Dong, F. Zhang and J. Jin, Nano Lett., 2013, 13, 1711 CAS.
- P. Wu, N. Du, H. Zhang, C. Zhai and D. Yang, ACS Appl. Mater. Interfaces, 2011, 3, 1946 CAS.
- J. Qiu, P. Zhang, M. Ling, S. Li, P. Liu, H. Zhao and S. Zhang, ACS Appl. Mater. Interfaces, 2012, 4, 3636 CAS.
- Y. Fu and J. Shen, Chem. Commun., 2007, 2172 RSC.
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904 CrossRef CAS PubMed.
- B. Liu, A. Abouimrane, Y. Ren, M. Balasubramanian, D. Wang, Z. Z. Fang and K. Amine, Chem. Mater., 2012, 24, 4653 CrossRef CAS.
- S. J. Chang, J. B. Park, G. Lee, H. J. Kim, J. B. Lee, T. S. Bae, Y. K. Han, T. J. Park, Y. S. Huh and W. K. Hong, Nanoscale, 2014, 6, 8068 RSC.
- X. Pan, Y. Zhao, G. Ren and Z. Fan, Chem. Commun., 2013, 49, 3943 RSC.
- G. Ji, Y. Ma, B. Ding and J. Y. Lee, Chem. Commun., 2012, 3329 CAS.
- Z. Xu, H. Wang, Z. Li, A. Kohandehghan, J. Ding, J. Chen, K. Cui and D. Mitlin, J. Phys. Chem. C, 2014, 118, 18387 CAS.
- Y.-J. Bai, C. Gong, N. Lun and Y.-X. Qi, J. Mater. Chem. A, 2013, 1, 89 CAS.
- S.-L. Chou, J.-Z. Wang, H.-K. Liu and S.-X. Dou, J. Phys. Chem. C, 2011, 115, 16220 CAS.
- G.-N. Zhu, Y.-G. Wang and Y.-Y. Xia, Energy Environ. Sci., 2012, 5, 6652 CAS.
- Z. Jian, L. Zhao, R. Wang, Y.-S. Hu, H. Li, W. Chen and L. Chen, RSC Adv., 2012, 2, 1751 RSC.
- Y. M. Jiang, K. X. Wang, X. Y. Wu, H. J. Zhang, B. M. Bartlett and J. S. Chen, ACS Appl. Mater. Interfaces, 2014, 6, 19791 CAS.
- C. C. Li, Q. H. Li, L. B. Chen and T. H. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 1233 CAS.
- A. Nugroho, S. J. Kim, K. Y. Chung, B.-W. Cho, Y.-W. Lee and J. Kim, Electrochem. Commun., 2011, 13, 650 CrossRef CAS PubMed.
- K. Mukai, Y. Kato and H. Nakano, J. Phys. Chem. C, 2014, 118, 2992 CAS.
- Y. Q. Wang, L. Gu, Y. G. Guo, H. Li, X. Q. He, S. Tsukimoto, Y. Ikuhara and L. J. Wan, J. Am. Chem. Soc., 2012, 134, 7874 CrossRef CAS PubMed.
- H. Ni, W.-L. Song and L.-Z. Fan, Electrochem. Commun., 2014, 40, 1 CrossRef CAS PubMed.
- J. M. Feckl, K. Fominykh, M. Doblinger, D. Fattakhova-Rohlfing and T. Bein, Angew. Chem., Int. Ed., 2012, 51, 7459 CrossRef CAS PubMed.
- Y. Sha, B. Zhao, R. Ran, R. Cai and Z. Shao, J. Mater. Chem. A, 2013, 1, 13233 CAS.
- J. Haetge, P. Hartmann, K. Brezesinski, J. Janek and T. Brezesinski, Chem. Mater., 2011, 23, 4384 CrossRef CAS.
- J. Ma, C. Wang and S. Wroblewski, J. Power Sources, 2007, 164, 849 CrossRef CAS PubMed.
- A. D. C. Permana, A. Nugroho, K. Y. Chung, W. Chang and J. Kim, Chem. Eng. J., 2014, 241, 216 CrossRef CAS PubMed.
- Y. Wang, H. Xia, L. Lu and J. Lin, ACS Nano, 2010, 4, 1425 CrossRef CAS PubMed.
- L. Gao, R. Liu, H. Hu, G. Li and Y. Yu, Nanotechnology, 2014, 25, 175402 CrossRef PubMed.
- J. Chen, L. Yang, S. Fang, S.-I. Hirano and K. Tachibana, J. Power Sources, 2012, 200, 59 CrossRef CAS PubMed.
- A. Nugroho, S. J. Kim, W. Chang, K. Y. Chung and J. Kim, J. Power Sources, 2013, 244, 164 CrossRef CAS PubMed.
- J. Huang and Z. Jiang, Electrochem. Solid-State Lett., 2008, 11, A116 CrossRef CAS PubMed.
- Q. Zhou, L. Liu, H. Guo, R. Xu, J. Tan, Z. Yan, Z. Huang, H. Shu, X. Yang and X. Wang, Electrochim. Acta, 2015, 151, 502 CrossRef CAS PubMed.
- C. Wang, Z.-X. Deng and Y. Li, Inorg. Chem., 2001, 40, 5210 CrossRef CAS PubMed.
- C. Lin, X. Fan, Y. Xin, F. Cheng, M. O. Lai, H. Zhou and L. Lu, Nanoscale, 2014, 6, 6651 RSC.
- L. Shen, C. Yuan, H. Luo, X. Zhang, K. Xu and Y. Xia, J. Mater. Chem., 2010, 20, 6998 RSC.
- Z. Xu, Z. Li, C. M. B. Holt, X. Tan, H. Wang, B. S. Amirkhiz, T. Stephenson and D. Mitlin, J. Phys. Chem. Lett., 2012, 3, 2928 CrossRef CAS.
- H.-G. Jung, S.-T. Myung, C. S. Yoon, S.-B. Son, K. H. Oh, K. Amine, B. Scrosati and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 1345 CAS.
- L. Sun, J. Wang, K. Jiang and S. Fan, J. Power Sources, 2014, 248, 265 CrossRef CAS PubMed.
- Y.-S. Lin and J.-G. Duh, J. Power Sources, 2011, 196, 10698 CrossRef CAS PubMed.
- W. Liu, J. Zhang, Q. Wang, X. Xie, Y. Lou, X. Han and B. Xia, Powder Technol., 2013, 247, 204 CrossRef CAS PubMed.
- Z. He, Z. Wang, F. Wu, H. Guo, X. Li and X. Xiong, J. Alloys Compd., 2012, 540, 39 CrossRef CAS PubMed.
- Y.-S. Lin, J.-G. Duh, M.-C. Tsai and C.-Y. Lee, Electrochim. Acta, 2012, 83, 47 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- Z. Li, Z. Xu, X. Tan, H. Wang, C. M. B. Holt, T. Stephenson, B. C. Olsen and D. Mitlin, Energy Environ. Sci., 2013, 6, 871 CAS.
- B. Key, D. J. Schroeder, B. J. Ingram and J. T. Vaughey, Chem. Mater., 2012, 24, 287 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra03158c |
| This journal is © The Royal Society of Chemistry 2015 |