
Photoreduction of natural redox proteins by CdTe quantum dots is size-tunable and conjugation-independent†
Abstract
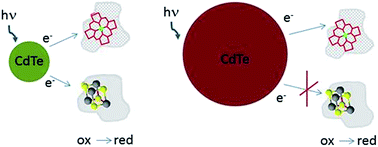
Colloidal CdTe quantum dots (QD) were able to reduce both heme and iron–sulfur cluster containing proteins. Reduction was depended on quantum dots size. CdTe nanocrystals emitting light with maximum at 550 nm (QD-550, r ∼ 1.5 nm) reduced both ferredoxin (Fd) and cytochrome c (Cyt c). The process was followed by UV/Vis absorption spectroscopy and steady state fluorescence spectroscopy. For CdTe emitting longer wavelength (QD-610 and QD-670) reduction of Fd was less efficient. QD emitting light with maximum at 750 nm (QD-750, r ∼ 3.3 nm) reduced only Cyt c. Reduction of proteins by QDs was photo-dependent and did not demand oxygen presence. As shown by gel filtration and fluorescence correlation spectroscopy, Fd formed complexes with QD while Cyt c did not bound steadily. The stable complex was not necessary for photoreduction, although might influence its kinetics. Time-resolved fluorescence studies showed than electron transfer rate depends on QD size and also is higher for QD–Cyt c when compared to QD–Fd. The mechanism of process was additionally explored by detailed analysis of QD–protein complex formation and by measurements of cyclic voltamperometry and zeta potential. These results open new possibilities in controlling of natural redox processes.
 Please wait while we load your content...
Please wait while we load your content...

