
Magnesium-based systems for carbon dioxide capture, storage and recycling: from leaves to synthetic nanostructured materials†
Jenny G. Vitillo
*
Department of Science and High Technology, Università dell'Insubria, Via Lucini 3, 22100 Como, Italy. E-mail: jenny.vitillo@uninsubria.it; Fax: +39 031 2386630; Tel: +39 031 2386614
First published on 8th April 2015
Abstract
A steep rise of carbon dioxide level in the atmosphere is one of the main causes of global warming. This increase is ascribed to the fact that, since the beginning of the industrial revolution, natural processes for CO2 sequestration are no longer able to cope with the excess of CO2 produced by anthropogenic activities. In recent years, research has been focused on defining artificial CO2 cycles to support the natural one. The element magnesium is used in this review as leitmotif to explore the majority of systems involved in each step of the natural and artificial CO2 cycles (separation, storage, sequestration or recycling). Magnesium is in fact ubiquitous, being present in the mesosphere as global layers, on the Earth's surface in the most important enzyme for carbon fixation (Rubisco) and in silicates that constitute the major part of rocks, where CO2 is sequestered through natural weathering. For what concerns synthetic materials, zeolites, metal-supported particles and metal–organic frameworks are only a few of the systems considered in the literature. The intent of this review is to connect different fields of study to create an interdisciplinary review in the chemistry domain. Research outlooks are suggested for the different fields. In the end, a qualitative analysis of the advantages and limits of different processes and a rough estimate of their potential are given in terms of the time needed to reduce the atmospheric CO2 level. Although economical, political and health evaluations would be also necessary, this analysis indicates that forestation could be a possible winning solution in the short-middle term for lowering the atmospheric CO2 concentration.
 Jenny G. Vitillo | Jenny G. Vitillo received her PhD in Materials Science in 2005 from the University of Torino (Italy). She held a post doctoral position at the same university with the Physical Chemistry group up to 2012, where she improved her experimental/computational skills. In 2013, after receiving financing for a FIRB project (Italian starting grant), she moved to the University of Insubria (Italy) with a Post doc fellowship. Her research is focused on three main subjects and in particular light harvesting through dye/nanoscaffold systems, hydrogen storage and, last but not least, carbon dioxide capture and recycling. |
1. Introduction
Carbon dioxide is the most abundant greenhouse gas in the terrestrial atmosphere. Its presence keeps the Earth from being a cold rock in space. Moreover, CO2 is also of paramount importance being at the basis of essential processes such as photosynthesis. Since 1750 (that is at the beginning of the Industrial Revolution) a steep and continuous increase of its atmospheric level has occurred, after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 years of constant concentration (see Fig. 1).1,2 This phenomenon has a well established anthropogenic origin and can be mainly related to fossil fuels combustion (73.8%), agriculture and deforestation (22.6%) and other sources (3.6%).3 In 265 years, CO2 concentration in the atmosphere increased from pre-industrial 280 to the present day 400 ppmv, corresponding to an excess of about 9 × 1014 kg of CO2 (900 metric gigatons).4 It is noteworthy that the last part of the plot reported in Fig. 1b shows an almost linear dependence of CO2 level with time. If a linear fitting of those data is performed, the following relationship is obtained:
000 years of constant concentration (see Fig. 1).1,2 This phenomenon has a well established anthropogenic origin and can be mainly related to fossil fuels combustion (73.8%), agriculture and deforestation (22.6%) and other sources (3.6%).3 In 265 years, CO2 concentration in the atmosphere increased from pre-industrial 280 to the present day 400 ppmv, corresponding to an excess of about 9 × 1014 kg of CO2 (900 metric gigatons).4 It is noteworthy that the last part of the plot reported in Fig. 1b shows an almost linear dependence of CO2 level with time. If a linear fitting of those data is performed, the following relationship is obtained:| [CO2] = 17.084t − 2615.0 |
000 years such a high value was never reached, being [CO2] values always lower than 300 ppmv.1 Fig. 1a also reports the surface temperature profile of the past 420000 years in Antarctica, as in ref. 1. The temperature values are referred to the average temperature in 1950. A strong and direct correlation between the concentration of CO2 and the temperature is evident,1,5 with a short delay between the variation in [CO2] and the corresponding change in temperature ΔT. Linear fitting of those data gives the phenomenological equation:| ΔT = 0.092[CO2] − 25.7 |
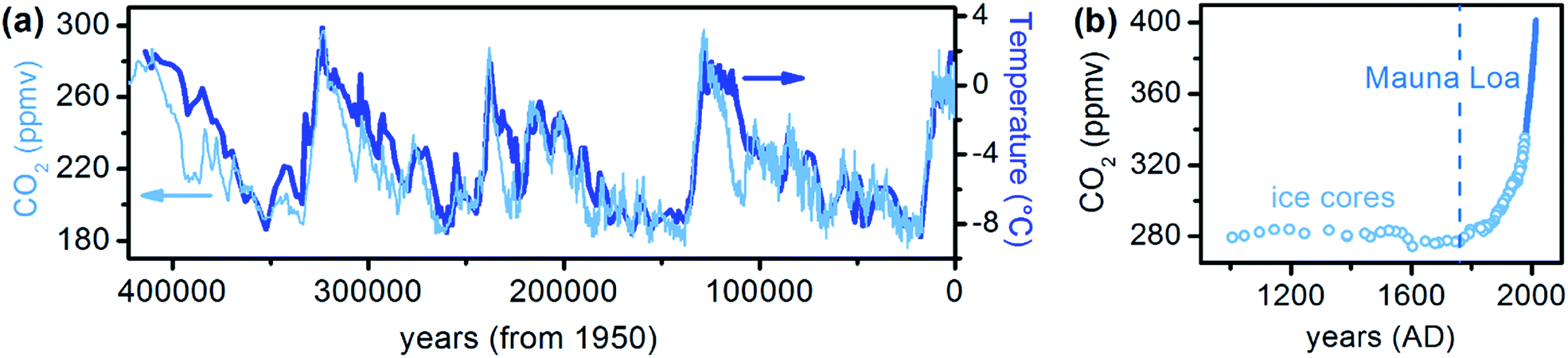 | ||
| Fig. 1 (a) Atmospheric CO2 profile and surface temperature change with respect to the value recorded in 1950 in the past 420000 years in Antarctica, accordingly to the Vostok ice cores recording. Data as reported in ref. 1. (b) Atmospheric CO2 concentration dependence with time accordingly to data recorded on Vostok ice cores1 (light blue circles) and directly measured from atmosphere at the Mauna Loa observatory (blue solid line).2 The perfect accordance between the two sets of data is evident. The dashed vertical line indicates the beginning of the industrial revolution. | ||
More precise climate model simulations, although differing in the absolute value of ΔT, all accordingly predict an increase in the planet temperature, as already occurring. Contemporaneously, other related global changes have been verified in a cause-effect cascade, e.g. the raise in global sea level (3.18 ± 0.4 mm per year),6 and an increase in the frequency of extreme weather events (floods, hurricanes, …).7 It is evident that a further increase in carbon dioxide concentration would make these events even more drastic. On the other hand, the immediate stop of the anthropogenic CO2 emissions would require a drastic technological downgrade that is neither possible, nor acceptable. In order to help in the visualization of the enormity of the CO2 amount already present in excess in the atmosphere, three different pictorial representations are reported in Fig. 2 (see ESI† for details). This amount would correspond to about: (i) 255000 bottles of liquid CO2 having the size of the Empire State Building (Fig. 2a); (ii) the CO2 fixed in the wood of a forest covering the entire Earth surface (Fig. 2b); (iii) twenty mountains of magnesium carbonate having the size of Monviso (Fig. 2c). It is evident that just the capture of this amount would represent a pharaonic project. The ineluctable dependence of humankind from fossil fuels which is likely to continue in the future decades makes the prospect even worse. Adapt, mitigate or ignore? was the provocative question made by Sir David King in one of its famous articles,7 as the three possible alternative in front of a problem that seems without an easy solution. Following the need to mitigate, the research community is deeply engaged in the search for substitute energetic routes or more efficient energy management aimed to lower or eliminate the equation implicit in the use of fossil fuels: energy = CO2 emission. At the same time, research addresses the definition of efficient artificial flows in the CO2 cycles, aimed to support the natural ones (see Scheme 1).
 | ||
| Fig. 2 Pictorial representations aimed to help in the visualization of the excess amount of CO2 present in the atmosphere in 2015: (a) 255000 bottles of CO2 liquid having the size of the Empire State Building; (b) the CO2 fixed in the wood of a two years old forest covering the entire Earth surface; (c) twenty mountains of magnesium carbonate having the size of Monviso (height 3842 m). | ||
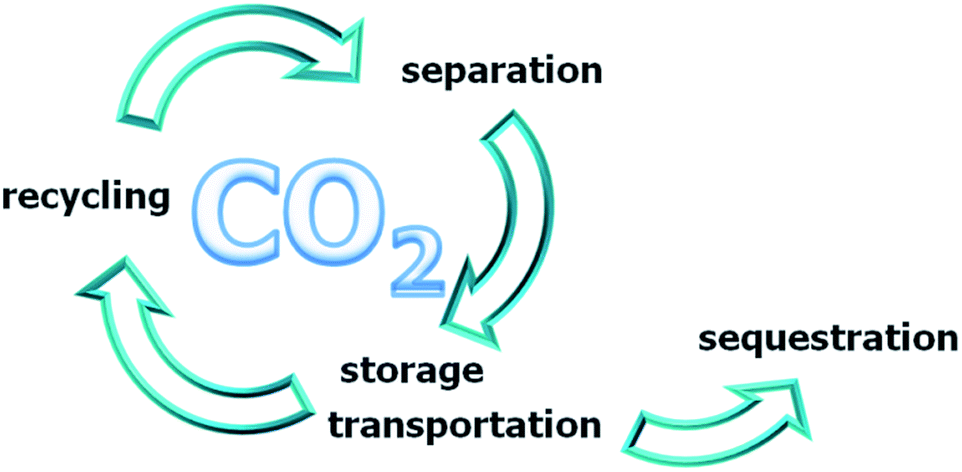 | ||
| Scheme 1 CO2 cycle. | ||
This artificial CO2 cycle is constituted by three steps: (i) separation, where CO2 is separated from air8 or from flue gases of stationary point sources (e.g. coal-fired power plants); (ii) short term storage and/or transportation of CO2;9 (iii) long term storage (sequestration) in general in a subterranean or submarine storage site (CCS process).10 However, an excess of 900 Gtons of CO2 can be also taken as an opportunity, considering carbon dioxide as a C1 feedstock, cheap and everywhere abundant. CO2 recycling (CCU process), as an alternative to sequestration, constitutes an appealing strategy, allowing the transformation of a “problem” in valuable chemicals, e.g. the transformation of CO2 in fuels. It is evident that it is mandatory in this case differentiating the number of possible CO2-based reactions (and then of products). In fact, the amount of available CO2 is so large that any single chemical would fast saturate the market.11 This is a very important research field, being at present only a small fraction of the CO2 recycled.11–16
It is crucial to keep in mind that the amount of the materials that will be employed significantly in the CO2 cycle would have to be approximately of a similar order of magnitude of the excess of CO2 itself. Systems composed in large part of rare or costly elements will not be applicable on a global scale. Magnesium is the fourth most common element in the Earth's crust, making up 13.9% of the planet mass. It is then a good choice as fundamental constituent for materials to be used with this aim. In literature, magnesium based systems have been reported for each of the CO2 cycle steps. Some of them showed superior performances with respect to any other material of their class, especially in applications that require a high affinity of the material for CO2, such as gas separation and catalysis. It should be noted that Rubisco, the enzyme at the basis of the most important carbon dioxide recycling process on Earth, that is carbon fixation in plants, is a Mg-based enzyme.
In the following, a general description of the CO2 molecule and of the different strategies adopted in CO2 separation, sequestration and recycling will be presented. Mg-based representatives of the different classes of materials will be reviewed along with their applications in the different steps of the CO2 cycle: enzymes, Grignard reactants, metal–organic frameworks (MOFs), metal surfaces, Mg–O based oxides, zeolites, clays, hydroxides, silicates, and hydrides.
For each of these systems, a description of their structure and of the mechanism and energetics of the interaction with CO2 will be presented, when possible, along with a review of their performances. The factors at the basis of the suitability of Mg2+ materials for interaction with CO2 will be also discussed. In the conclusions, outlooks on the future research on materials have been reported. A qualitative analysis of the systems proposed for the capture of atmospheric CO2 is also shown, with the aim to individuate a possible solution for the mitigation of CO2 level in the short-middle term.
2. CO2
2.1. The CO2 molecule
Carbon dioxide is a linear molecule characterized by a significant quadrupolar moment (see Table 3). Its electrostatic potential can be qualitatively described like a positive doughnut in the C plane with two negative lobes on the molecular axis (see Fig. 9a).17 CO2 can then act both as a Lewis acid and Lewis base on pure electrostatic basis, interacting in a side-on geometry through a direct interaction with C with negative charges,18 whereas in the case of positive charges end-on adducts through one O are formed.17 However, experiments and calculations converge in indicating that adsorption is a complex interplay of dispersion forces and electrostatics, favoring cooperative adsorption based on the interaction through both the C and O atoms with the material. This means that CO2 adsorption and activation are favored on surfaces where strong Lewis acid–base couples are present, as for example on oxide surfaces. It is then evident that favoring intermolecular interactions between CO2 molecules (that is between quadrupoles) is beneficial on the CO2 capacity of materials.19Several geometries of coordination on surfaces are possible (see for example Fig. 12 and Section 4.6 for further details), which nature can determine different reaction paths. These species can be identified through the use of spectroscopies, such as 13C nuclear magnetic resonance (NMR),20,21 and infrared (IR) spectroscopies.22 For what concerns IR spectroscopy, carbon dioxide is characterized by three vibrational modes: the IR-inactive symmetric stretching mode ![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/i_char_e0e1.gif) 1 (1388.3 cm−1 for 12CO2 gas),23 the doubly degenerated bending vibration 2 (667.3 cm−1)23 and the asymmetric stretching mode 3 (2349.3 cm−1).23 Upon interaction with the materials, these modes are only slightly shifted, even when highly charged species are present.23 The shift of the CO2 vibrations upon adsorption gives some insight into the adsorption geometry and the nature of the adsorption site. The presence of a negative charge, although leaving identical (and elongated) the two C
1 (1388.3 cm−1 for 12CO2 gas),23 the doubly degenerated bending vibration 2 (667.3 cm−1)23 and the asymmetric stretching mode 3 (2349.3 cm−1).23 Upon interaction with the materials, these modes are only slightly shifted, even when highly charged species are present.23 The shift of the CO2 vibrations upon adsorption gives some insight into the adsorption geometry and the nature of the adsorption site. The presence of a negative charge, although leaving identical (and elongated) the two C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds, causes a change in the ∠OCO angle: this corresponds to the decrease of the mode and to the removal of the degeneracy for the mode. On the contrary, in the case of adsorption on a positive charge the molecule is still linear but the two CO bond are no more identical: the C–O bond adjacent to the cation lengthens slightly whereas the other C–O bond shortens of almost the same amount.23 This spectroscopically corresponds to an increase of the asymmetric stretching mode frequency.24 It is important to remind at this point that if the adsorption in a microporous material is considered, a strong matrix confinement effect is observed on the vibrational modes of CO2 because of its significant quadrupole moment.23 A shift in the mode to 2338 cm−1 has been estimated for the unperturbed molecule in microporous materials due to confinement.17 If chemical bonded species are formed, signals in the 1800–800 cm−1 region are observed, the position of which bands is indicative not only of the nature of the formed species but also of the geometry of coordination.12,22 A comprehensive review of the characterization by infrared spectroscopy of CO2 surface species on oxide surfaces is reported in ref. 22.
O bonds, causes a change in the ∠OCO angle: this corresponds to the decrease of the mode and to the removal of the degeneracy for the mode. On the contrary, in the case of adsorption on a positive charge the molecule is still linear but the two CO bond are no more identical: the C–O bond adjacent to the cation lengthens slightly whereas the other C–O bond shortens of almost the same amount.23 This spectroscopically corresponds to an increase of the asymmetric stretching mode frequency.24 It is important to remind at this point that if the adsorption in a microporous material is considered, a strong matrix confinement effect is observed on the vibrational modes of CO2 because of its significant quadrupole moment.23 A shift in the mode to 2338 cm−1 has been estimated for the unperturbed molecule in microporous materials due to confinement.17 If chemical bonded species are formed, signals in the 1800–800 cm−1 region are observed, the position of which bands is indicative not only of the nature of the formed species but also of the geometry of coordination.12,22 A comprehensive review of the characterization by infrared spectroscopy of CO2 surface species on oxide surfaces is reported in ref. 22.
First step in reactions involving CO2 is its activation, that in all cases asks for the formation of a CO2− anion with a change from the linear to a bent geometry.12 More precisely, dissociation of CO2 likely happens through the formation of a Freund–Messmer anionic complex (CO2−⋯CO2), that is through an intermediate CO2δ− state. The factors that influence the formation of chemisorbed CO2 species are: (i) the electronic configuration of the system; (ii) its work function (as a measure for the ease electron transfer); (iii) the influence of local geometry.12 The change in the CO2− geometry from the linearity of the neutral CO2 can be explained by considering the qualitative scheme reported in ref. 12, showing the molecular orbitals of CO2 for both the linear and bent configurations (Fig. 3). The HOMO (light blue) and the LUMO (green) orbitals are indicated with colors to facilitate their identification. It is clear that moving from linear to bent configuration, π-orbitals show the most pronounced alterations in energies with a split of all the degeneracies.12 The perturbation is particularly important for HOMO and LUMO: upon bending, the inversion in stability of one of LUMO orbitals (the 2πu-6a1 one) with respect to the HOMO ones is observed. The occupation of this 2πu-6a1 orbital by the electron transferred from the surface determines the bond angle.12 Gas phase measurements indicate that the average C–O bond order in CO2− is only 1.5 as compared to 2 in neutral CO2.12 This is in line with the larger C–O distance (1.24 versus 1.15 Å in neutral CO2) and the smaller bond enthalpy of C–O in CO2− with respect to CO2, making the anionic form more unstable upon dissociation.12 The formation of this anionic species is a strong rate determining step in reactions involving CO2. The potential associated to the reaction:
CO2 + e− ![[left over right harpoons]](https://www.rsc.org/images/entities/char_21cb.gif) CO2˙− CO2˙− |
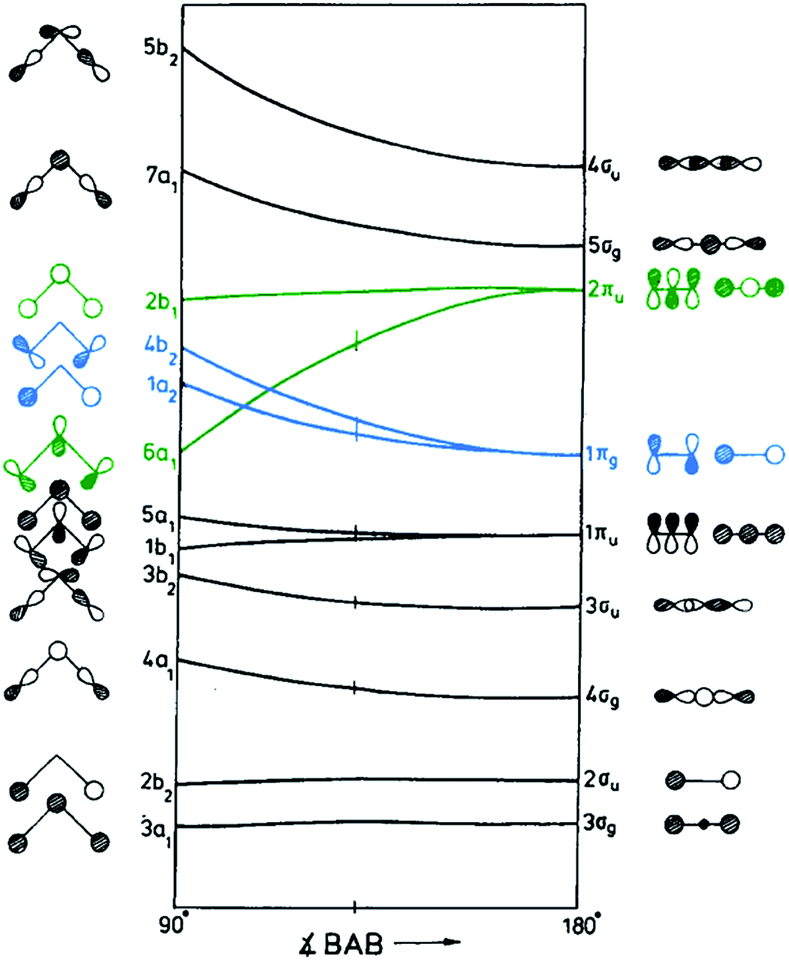 | ||
| Fig. 3 Walsh diagram of CO2 orbital energies in linear (right) and bent geometries (left). HOMO and LUMO levels have been signalized by light blue and green colors, respectively. Adapted from ref. 12. Reproduced with permission. Copyright Elsevier 1996. | ||
2.2. CO2 recycling
The use of CO2 as chemical feedstock was12 in 1996, and still nowadays is limited to few industrial processes:14 synthesis of urea and its derivatives, salicylic acid and carbonates. Only 128 Mt per year of CO2 are in fact utilized in industry.14 This low exploitation of CO2 as reagent is due to two main factors: the low efficiency of the catalysts and the high CO2 cost. In fact, although it is in general claimed in academic literature that CO2 is a highly abundant and cheap reactive, it is important to remind that it is rarely present as pure in nature and the separation cost is energetically demanding.15 Actually, CO2 is expensive because its separation cost is high and this is the main reason for the absence of a particular interest on CO2 on the industrial point of view.15 Nevertheless the CO2 emission restrictions due to Kyoto protocol can push towards the use of CO2 as reagent, by creation of a market for the separated CO2.In addition to that, CO2 is often described as a molecule with a low reactivity. This is not always the case, being for example, a selective oxidant towards hydrocarbons.15 Moreover, most of the reactions are favorable on the thermodynamic point of view (see Table 1: the entropic term gives a little contribution to the Gibbs free energy so that ΔH is a good guide to thermodynamic feasibility of those reactions).12 The reason of the low reactivity reputation has a kinetics origin and relies on the high energy needed for the formation of the intermediate anionic species.12 Additionally, if the reaction requires the breaking of a C–O bond, substantial energy has to be supplied for its cleavage,12 making the activation of CO2 the main rate determining step in both natural and artificial processes.
| Reactions | Name | ΔHa (kJ mol−1) | Ref. | n. |
|---|---|---|---|---|
| a Reaction enthalpies at 298 K. | ||||
| CO2 + H2 CO + H2O (g) |
Reverse water gas shift (RWGS) | +41.2 | 14 | [1] |
| CO2 + H2 CO + H2O (l) |
−2.8 | 12 | [1]bis | |
| CO2 + 4H2 CH4 + 2H2O |
Methanation of carbon dioxide or Sabatier reaction | −164.9 | 26 | [2] |
| CO2 + 3H2 CH3OH + H2O |
Synthesis of methanol | −49.5 | 14 | [3] |
| CO2 + H2 HCOOH |
Formic acid synthesis | −31.8 | 27 | [4] |
| CH4 + CO2 2CO + 2H2 |
Dry reforming of methane | +247 | 28 | [5] |
| CH4 + CO2 CH3COOH |
Carboxylation of methane | −16.6 | 15 | [6] |
| C6H6 + CO2 C6H5COOH |
Carboxylation of benzene | −40.7 | 15 | [7] |
| 2MeOH + CO2 CH3COCH3 + H2O |
Carboxylation of methanol | −27.9 | 29 | [8] |
| Mg(OH)2 + CO2 MgCO3 + H2O |
Carbonation | −19.7 | 30 | [9] |
| MgO + CO2 MgCO3 |
Carbonation | −100.9 | 30 | [10] |
| MgSiO3 + CO2 MgCO3 + SiO2 |
Carbonation of pyroxene | −81 | 31 | [11] |
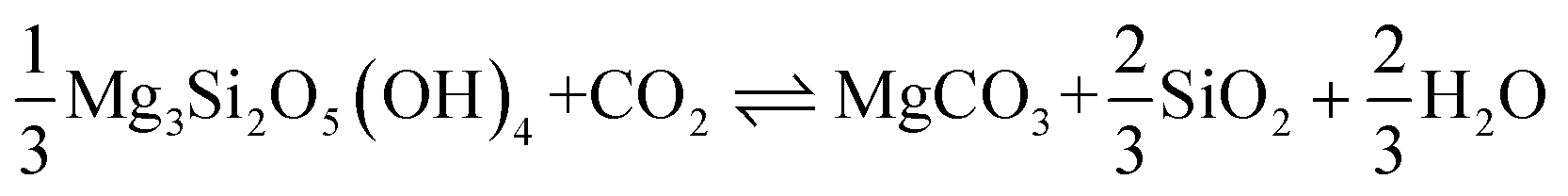 |
Carbonation of chrysotile | −35 | 31 | [12] |
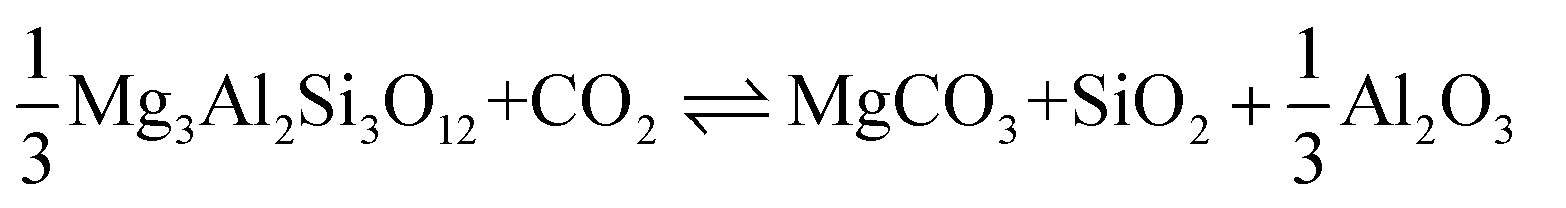 |
Carbonation of pyrope (garnet) | −92 | 31 | [13] |
All the reactions involving CO2 can be divided in two groups: (ia) reactions where CO2 is inserted as a whole in the products and (iia) reactions that require to break one or more carbon–oxygen bond. Another classification, only in part coincident with the previous one, separates the reactions between (ib) reactions leaving unchanged the oxidation state of carbon and (iib) reactions where CO2 reduction is required. In both cases, the second group of reactions asks more energy than the first group. It is interesting to notice that the most important reactions aimed to carbon fixation in nature, that is the ones catalyzed by Rubisco and PEPC enzymes, belong to the (ia) group (see Section 4.1). Moreover, in general larger the change in the oxidation state, larger the activation energy (Ea), and then slower kinetics of reaction are expected.15 For example, in methanation reaction (that requires the largest change in the oxidation state of carbon from +4 to −4) an Ea of 106.9 ± 0.5 kJ mol−1 has been reported for the reaction catalyzed by Pd/Mg–SiO2,32 whereas CO formation (change from +4 to +2) has an apparent activation energy of only 52.7 ± 0.2 kJ mol−1. This last statement is not always true: for example in gas phase complexes of CO2 with Ti, the insertion of the metal in one CO bond was observed without energetic barrier.33 CO2 is also dissociatively chemisorbed with the formation of surface carbide and oxide on Mo surfaces already at RT.12
Among all the possible reactions involving CO2,13,15 the discussion will be restricted in the following to hydrogenation reactions (reactions [1]–[4] in Table 1), methane reforming (reaction [5]), carboxylation of methanol (reaction [8]) and inorganic carbonation (reactions [9]–[13]). It is important to stress that the reactions involving CH4/CO2 and CO2/H2 mixtures can represent alternatives to separation processes, when possible.
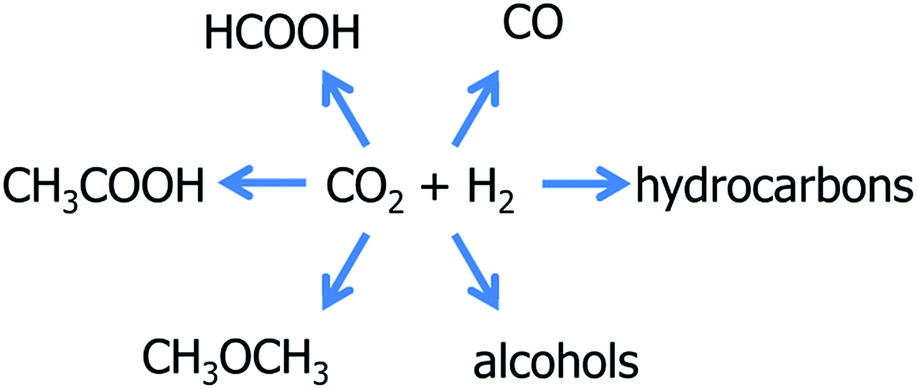 | ||
| Scheme 2 Hydrogenation reactions. | ||
The formation of one among all the possible products is obviously determined by the CO2/H2 ratio in the gas feed and, because of the different reaction enthalpies (see Table 1), by the temperature and pressure conditions adopted. Additionally, the nature of the catalyst and any detail of its composition and preparation (calcination procedure, reduction temperature) determine the formation of one or another reaction product (see Section 4.6).14 This sensitivity can represent a big advantage for the CO2/H2 systems: in principle it is possible to vary the reaction path and then the products by small changes in the reaction conditions. Also the presence of small pollutants in the gas feed can be determinant on the reaction path.14 Nevertheless, this also clearly indicates that each of these reactions can constitute a side reaction for each of the others. Both homogeneous and heterogeneous catalysts have been reported to be active in CO2 hydrogenation,14 allowing the formation of very different reaction products. Among the heterogeneous catalysts, metals, crystalline oxides (perovskite and pyroclores) and metal particles supported on oxides and carbons have been reported. Although for all these reactions, the reaction mechanism is nowaday unknown, the combination of the results reported so far in literature allows to identify some common points, that are here summarize in the following ten bullet points.
(i) The rate determining step in all these reactions is the CO2 activation. For example, activation energies for reactions [1] and [2] on Ni(100) surface have very similar values:34 88.7 for [2] and 72.8–82.4 kJ mol−1 for [1]. This allows to suppose that their rate determining step is common. Moreover, hydrogenation/dehydrogenation on metal particles is known to be a relatively fast process. The mobility of the H species on the surface will determine the formation of methane or CO. The reaction would proceed to the formation of methane if hydrogen is fast dissociated and can move easily on the surface towards the carbonate species (and/or vice versa).35 On the contrary CO is released (see Sections 4.6.2 and 4.9).
(ii) Catalysts for these reactions are in almost the totality of the cases, metal particles supported on oxides, where the metals are transition metals active in hydrogen splitting.
(iii) On supported metal particle catalysts, the reaction is a dual site one if the oxidic support is acidic or basic in nature, whereas it is single site one if the support is inert (e.g. SiO2, carbon). In dual site reactions, hydrogen is dissociated on the metal particles, whereas CO2 is activated on the oxidic system through the formation of formate (acidic oxides) or carbonate/carboxylic species (basic oxides). The recombination of the two units happens at the boundary between the metal particle and the oxide surface. The metal particles participate significantly to the decomposition of CO2 only if supported on inert support and if they are suitable for doing it: e.g. Rh is able to activate CO2, whereas Ni and Pt are not.28
(iv) Ni, Cu and Fe-based catalysts are the most widely used catalysts in industry, although they suffer of a large sensitivity to impurities in the gas feed and to side products. Noble metals are on the contrary more active and characterized by a larger stability but their high cost makes impractical their large use. This means that, although largely academical studied, their large application on the industrial point of view would be hindered by economical issues.
(v) Addition of small amount of basic ions as promoters to the catalyst composition facilitates the activation of CO2 and reduces coke formation.
(vi) The catalyst preparation strongly influences the dimension and distribution of the metal particles, parameters that have a direct influence on the reaction path. To truly compare the different supports and metals by using literature results, attention have to be paid to the different preparation methods that can originate different aggregation of the metal particles making difficult or even impossible the evaluation.
(vii) Often a catalyst able to work for one of the hydrogenation reaction can be used for another.
(viii) High dispersion of the metal particles is suitable to enhance reaction kinetics. Nevertheless, dimensions lower than 100 Å have to be avoided because they can be more easily poisoned by impurities in the gas feed and by coke formation.
(ix) Choice of the suitable support is very important in primis because it determines the particle size, composition and dispersion. CO chemisorption and EXAFS study36 on supported 0.5% Rh on TiO2, MgO, SiO2, MCM-41 and γ-Al2O3, showed that the metal dispersion strongly depends upon support, increasing in the order: TiO2 < La2O3 < CeO2 < ZrO2 < MgO < SiO2 < MCM-41 < γ-Al2O3. Moreover, in the same study a change in the oxidation state of Rh was observed from metallic to cationic on TiO2 and γ-Al2O3, respectively.36
(x) Supports with a high oxygen mobility are preferred because they facilitate coke oxidation.
Among all the hydrogenation reactions, the most important ones can be identified in the reverse water gas shift14 (RWGS, reaction [1] in Table 1), the methanation of CO2 (Sabatier reaction, [2]), the methanol synthesis [3]35 and hydrocarbon synthesis.
Sabatier reaction. The hydrogenation of CO2 can proceed through further steps of hydrogenation or can have as only product the fully hydrogenation product of CO2, that is methane. Sabatier reaction is a highly efficient and highly exothermic reaction (ΔH298 K = −165 kJ mol−1), when carried out at temperature lower than 200 °C in an industrial process.37 This because of catalysts that allow to efficiently overcome the kinetic limitations due to the change from +4 to −4 in the oxidation state of carbon. Research in this field is concentrated on how to increase the lifecycle of the largely used Ni-based catalysts, that are easily deactivated by the formation of stable carbonyls. The economic issue for the synthetic methane is however represented mainly by the hydrogen production cost.35 Nevertheless, the huge and increasing availability of methane from natural sources would make more valid (on the environmental point of view) to purify methane from these sources or to reform it than to synthetize it (see Sections 2.2.2 and 2.3). A concrete application of the Sabatier reaction could be instead to use it to increase the methane concentration in biogas by using hydrogen from renewables.35 Other applications are related to space program for Mars colonization, where the Sabatier reaction would allow the easy production of a fuel and water.14,38,39
Reverse water gas shift. RWGS, being an endothermic reaction,14 is often present as side reaction wherever CO2 and H2 are present together in the reaction mixture. Its efficiency increases at high temperature (about 600 °C) or in presence of highly exothermic reactions, causing the poisoning of the catalysts, e.g. of Ni supported catalysts in methanation reactions. The most widely used catalysts in RWSG are copper based catalysts. However, Cu particles have a strong tendency to sinter, particularly at high temperature, easily causing the deactivation of the catalysts. Addition of small amounts of other metals (e.g. Fe) has shown to increase the stability of the catalysts, also up to 120 h.14
Synthesis of methanol. Methanol is a fuel and a very versatile reagent. Synthesis of methanol from CO2/H2 gas mixture can be obtained through the use of low reaction temperatures (250–350 °C) and high pressures.14 This reaction has in general a low selectivity being accompanied by the formation of almost all the other possible products.40 Cu/ZnO catalysts are considered the most effective catalyst for methanol formation, although the low activity and stability of Cu particles create major problems for practical applications as observed for RWGS. For this reason, new supports that allow to increase Cu particle stability (as perovskites) and the use of bimetallic metal particles are the directions followed in the research. New supports able to facilitate Cu reduction are also expected to increase the reaction yields.40 In fact, Cu is likely reduced to Cuα+ species during the reaction40 and this is thought to be a crucial step in the reaction mechanism.40 Formation of Cu+ would increase on the contrary the selectivity for the formation of CO, that is RWGS yields.40 For this reaction, beside Cu sintering, an important cause of deactivation is due to coke formation. Although outside the scope of the present review, it is important to stress as in the studies reported on these catalysts, the ability of ZnO to split hydrogen also at RT41 is often not taken into account.
Synthesis of hydrocarbons. Synthesis of hydrocarbons from CO2 is a two steps reaction that can involve CO or methanol as intermediates. In CO-mediated reactions, Co and Fe-based catalysts are the most widely used, whereas in methanol-mediated ones the catalysts adopted are the same used for methanol synthesis. For what concerns the CO-mediated synthesis, after CO formation the process continues as a classical Fisher Tropsch process. Nevertheless, direct hydrogenation of CO2 brings to the formation of very short chain hydrocarbons with a strong preference for methane. This is due to the slow adsorption rate of CO2 on the surface with a contemporaneous and fast dissociation and mobility of hydrogen, that favors the successive hydrogenation of intermediates.
| CH4 + CO2 2CO + 2H2 |
For this reason, in principle it can be directly applied to biogas, coal-seam, landfill gases or low quality natural gas, converting to syngas both the largest abundant greenhouse gases present (see Section 2.3). The syngas so obtained has in general a H2/CO ≤ 1. A syngas with this ratio increases the selectivity of Fischer–Tropsch towards long chain hydrocarbons, that is towards higher quality fuels.28 In fact if for the reaction:
| nCO + (2n + 1) H2 CnH2n+2 + nH2O |
| CO + H2O CO2 + H2 |
| 2nCO + (n + 1) H2 CnH2n+2 + nCO2 |
Although promising, dry reforming of methane suffers of several problems: (i) high presence of side reaction products (RWGS and coke formation); (ii) endothermicity of the reaction (ΔH298 K = +247 kJ mol−1); (iii) deactivation of the metal particles (due to sintering, reaction with the support, reaction with impurities in the gas stream, with oxygen and side reaction products). Among all these problems, coke formation is the major obstacle in dry methane reforming.28,42 Coke formation is predicted by thermodynamic models to cover a very wide temperature range (black curve in Fig. 4). The formation of this deposit can be due to different processes, as the Boudouard reaction:
| 2CO CO2 + C |
| CH4 C + 2H2 |
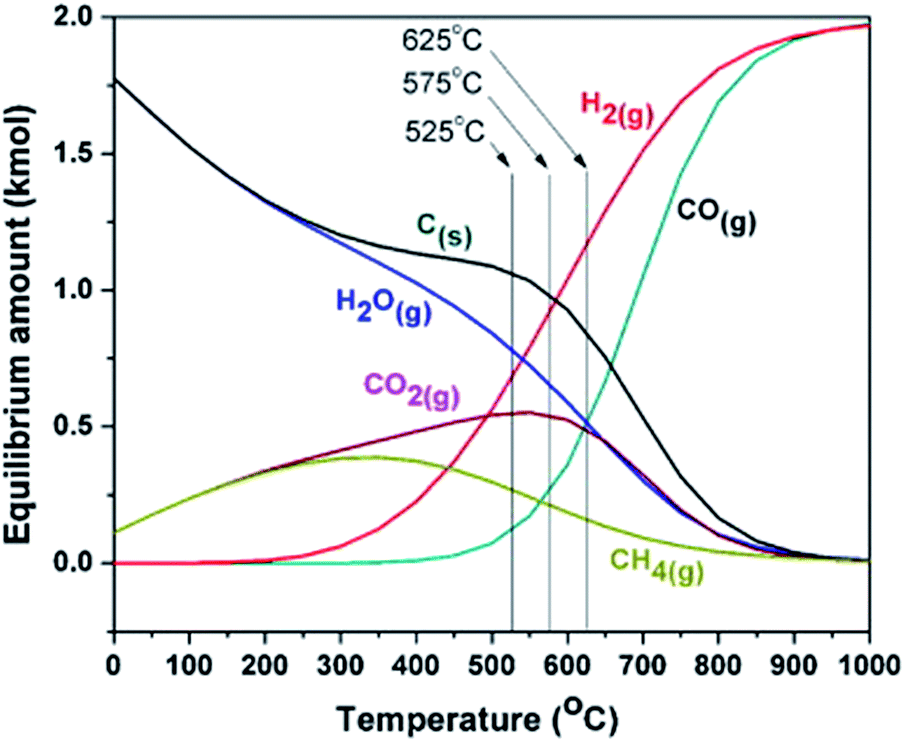 | ||
| Fig. 4 Thermodynamic equilibrium composition for dry methane reforming considering 1 kmol of CH4 and 1 kmol CO2 at 1 atm, from 0–1000 °C and at inlet feed ratio of CO2/CH4 = 1 and including the formation of coke. These plots were created by using Gibbs free energy minimization algorithm on HSC Chemistry 7.1 software. Reproduced with permission from ref. 44. Copyright 2013 Elsevier. | ||
All the considerations made in Section 2.2.1 for hydrogenation catalysts are also valid for the dry reforming ones. The main difference relies in the fact that the rate determining step in dry methane reforming is the adsorption and activation of methane, that is even less reactive than CO2 (see Table 3). In fact, the difference between the activation energies of CH4 and CO2 amounts to about 12 kJ mol−1 in several systems. Rh is the metal for which the dissociation of CH4 is most favorable, being observable from 150 °C, that is at significantly lower temperature than other transition metals.45 Also in this case, on a 1%Rh/MgO catalyst at 500 °C, Ea of 97 kJ mol−1 for CH4 and 85 kJ mol−1 for CO2 were obtained.28,45 Two mechanisms have been proposed for methane activation on metal surfaces: CH4 direct and indirect dissociation. In both cases, the formation of intermediates such as formyl or CHx species is hypothesized, with the oxygen coming then from the support (direct) or from the oxidant (indirect).28 In the first case, the surface oxygen would come from hydroxyls on acidic support and from carbonates on basic support.46 Likely both direct and indirect processes happen depending on the temperature, being the direct process favored for T > 550 °C.47 At T < 550 °C, CHx species can be formed as testified by the formation of C2H6.45 Activation of methane is carried out on the metal particles or at the interface between the metal and the support.28,48 Activation of CO2 happens exclusively on the support (if Rh catalyst are not used) whereas the recombination of the intermediates at the metal/support interface.28 In particular it has been proposed by pulse reactions, as CH4 would be dissociated in the first step of the reaction at the metal-oxide interface with formation of CO (in low amount) and H2 by leaving carbon species and oxygen vacancies on the support. The reaction would then proceed on the oxygen vacancies through CO2 dissociation to CO and oxidation of the left superficial carbon species to CO.49,50
As in hydrogenation reactions, also in this case the support plays a crucial role in the activation of CO2, important process for both the methane reforming reaction itself and for the coke removal.28 CO2 inert supports have then to be avoided for dry reforming of methane. Both acidic (formation of formate) and basic oxides (formation of carbonate/carboxylic/bicarbonate) have been successfully adopted. Solids characterized by high bulk oxygen conductivities and high thermal stabilities (>1000 °C), like perovskite materials, have shown to be highly suitable catalysts, actually bringing to an efficient oxidation of the coke formed in dry methane reforming.28
It is noteworthy that carbonate formation is an exothermic reaction (see reactions [9]–[13] in Table 1), where the amount of heat depends strongly on the reagent used. This heat can be a large fraction (up to 46% for60 CaO and 25% for MgO)61,62 of the heat released during the combustion process forming CO2 (393.8 kJ mol−1 CO2 for combustion of elemental carbon).31,60 This means that substantial heat is liberated in the overall chemical reaction, increasing the kinetics of the carbonation process. Moreover, for sufficiently fast processes, this heat can be efficiently recovered, further lowering the overall cost of the CCS process. For example, calcium looping technologies are at present one of the most efficient technologies for CO2 capture, although the very high temperature of reaction (T > 700 °C).63–65
Carbonation of dry carbonates has been also reported as a strategy to obtain chemical selective sorption of CO2 by maintaining a low regeneration temperature.56 In this case, separation process is carried out in presence of substantial amount of moisture in order to allow the formation of the corresponding bicarbonate species. Bicarbonates are in general less thermally stable than the corresponding carbonates, allowing the regeneration of the system at T < 200 °C. In literature, the most studied systems are K2CO3 and Na2CO3 that are CO2 sorbents with suitable adsorption temperatures in the 60–100 °C range with the possibility to fully regenerate the system at temperatures as low as 120–200 °C.63 Nevertheless, analogously to what observed for the other classes of materials, the carbonation yields are far from the stoichiometric amount.66 Dispersion on supports has been shown to be beneficial on the reaction yields.67
2.3. CO2 separation
CO2 separation processes are thought to be applied to four main gas mixtures: (i) air (N2/O2/CO2), (ii) natural gas (CO2/CH4), (iii) post-combustion flue gases (CO2/N2) and (iv) pre-combustion ones (CO2/H2). These separation processes differ significantly in the nature of the gases involved and in the CO2 partial pressure, besides in the temperature and (total) pressure conditions (see Table 2). Other separation processes (e.g. CO/CO2), having more restricted applications, would not be covered in this review.| Gas mixture | Composition (vol%)2,3,11,68 | T (°C) | P (bar) |
|---|---|---|---|
| Air | 78% N2, 21% O2, 0.04% CO2 | 25 | 1 |
| Natural gas | 30–60% CH4, 40–70% CO2 | >25 | 1–10 |
| Post-combustion | 70–77% N2, 15–16% CO2 | >50–75 | 1 |
| Pre-combustion | 61% H2, 35% CO2 | >40 | 30 |
The importance of atmospheric CO2 capture, although its high cost (not only on the energetic point of view), will be often stress in this review. Separation from air is decidedly the most challenging among those processes, being the concentration of CO2 very low (0.04%). Nevertheless, an efficient air capture would allow on one hand to mitigate the increase in the planet temperature and on the other, the existing fossil fuel based infrastructure to remain unaltered55 (at least until consumption of the fossil fuel stocks). Moreover, CO2 is three times as heavy as fuel55 and therefore cannot be stored in cars or airplanes. CO2 from these sources has then to be released in the atmosphere and recaptured later.55 Methane from natural sources (biogas, coal-seam, landfill gases, cow breeding) is rarely pure. The most important impurity (amounting also to the 60% on volume) is carbon dioxide. CH4 purification is then needed not only because CO2 decreases the heating value of methane but essentially because CO2 concentration higher than 5 vol% hinders even the catalytic oxidation of methane.68 Moreover, CO2 concentration in natural gas has to be below 3% in volume, in order to avoid pipeline corrosion. This restriction is less important existing several materials able to store high volumetric quantities of methane at RT.69,70 The huge and increasing availability of methane from natural sources and the important methane global warming potential (twenty-one time the CO2 one) make mandatory its use. In order to understand the amount of gas originating from these natural sources, in US the methane produced in landfill gas per year amount to about the 13–20% of total US CH4 production.28 Besides the possibility to chemically convert it in syngas (see Section 2.2.2), CO2/CH4 mixtures can be enriched in CH4 through the Sabatier reaction (by using renewable hydrogen, see Section 2.2.1)71 or by separation processes.
Post-combustion CO2 separations indicate that those processes can retrofit large stationary CO2 sources as coal-fired plants. In these case, flue gases resulting from the fuel combustion are predominantly containing N2 with CO2 concentrations of about 15%. Coal is currently the dominant fuel in the power sector and coal-fired plants are at present the most important CO2 sources. Any breakthrough system in the post-combustion class would be of paramount importance in the short middle term because it can retrofit the most largely abundant emission sites. Pre-combustion strategies, on the contrary, are thought to be applied to hydrocarbons/coal gasification plants, aimed to remove CO2 from H2-rich streams before energy conversion. This process, besides lowering CO2 emissions, enhances also the efficiency of the combustion. These processes are particularly interesting if applied to methane as in Integrated Gasification Combined Cycle plants (IGCC). It is important to stress as for what concerns post-combustion and pre-combustion processes, the temperatures reported in Table 2 are the lowest at which the separation can be carried out, being the ones at the smokestack exit. However, this does not imply that these are the most favorable temperatures if the efficiency of the whole process is taken into account. Efficient recovering of the heat evolved during the capture processes is in fact more difficult at these low temperatures. Post combustion separation processes are traditionally carried out by using aqueous solutions of amines, in particular monoethanolamine.11 These systems are characterized by high CO2 selectivities and capacities (0.5–1 mol CO2 per mol of amine, corresponding to about 2–4 mmol g−1)11 and by an intrinsic stability to moisture. Nevertheless, they present several problems, in primis the energy penalty that have to be paid for their regeneration, estimated as 25–40% in a coal-fired power station.11 Minimum energy required for CO2 separation in post-combustion systems would be only 3.5%, as estimated on pure thermodynamic basis.11 This large difference is in great part ascribed to the presence of water: regeneration of amines is in fact carried out at 100–140 °C. It is then evident that the most part of the heat provided would be spent for solvent heating and boiling. Amines have also other important drawbacks: these compounds are in fact costly, corrosive, carcinogenic and they easily undergo to thermal degradation.11
For these reasons, parallel to the research aimed to improve the performances of amine process, solid sorbents are deeply investigated in order to find a valid alternative as CO2 scrubbers. Because of the absence of solvents, solid sorbents have also the advantage of a wider temperature range of operation than liquid ones.63
Wang et al.59,63 have classified CO2 sorbents/scrubbers according to their working temperatures in (i) low temperature (<200 °C), (ii) intermediate temperature (200–400 °C) and (iii) high temperature (>400 °C) sorbents. They have also identified the important parameters to fully characterize a CO2 scrubber in: (i) working T and P conditions; (ii) working capacity (the actual reversible capacity of the system in the working conditions: see for example ref. 72); (iii) selectivity; (iv) durability; (v) kinetics; (vi) recycling stability; (vii) cost; (viii) regeneration cost.
In Table 3, the properties of the different gases involved in the separation processes reported in Table 2 are shown. For what concerns the geometrical point of view, CO2 has the smallest kinetic diameter with respect to all the other gases. Moreover, on the electrostatic point of view, CO2 would be characterized by the strongest interaction energy. Presence of high polar sites in materials would then increase their affinity towards the most polar molecule in the mixture (that is toward CO2) and then their selectivity.
| Gas | d (Å) | α (Å3) | μ (D) | Θ (D Å) | Tc (K) |
|---|---|---|---|---|---|
| CO2 | 3.30 | 2.507 | 0.000 | 4.30 | 304 |
| CH4 | 3.80 | 2.448 | 0.000 | 0.02 | 190 |
| N2 | 3.64 | 1.710 | 0.000 | 1.54 | 126 |
| H2O | 2.65 | 1.501 | 1.850 | 2.30 | 647 |
| CO | 3.76 | 1.953 | 0.112 | 2.04 | 133 |
On thermodynamic and molecular sieving bases, separation of CO2 from other gases would be then not a so difficult task in principle. Nevertheless, if high purity of CO2 after separation and low CO2 concentration in the gas feed are considered, this statement is no more true. Unfortunately, for CO2 recycling to different products from carbonates, high purity of the gas is requested.15 In this case, the production cost of CO2 is so high to limit its use as feedstock in industrial processes.15
The presence of water has to be also considered in real processes. It is evident from the properties reported in Table 3 that water presence can strongly lower the efficiency in sorption and separation of CO2. A quantum mechanical calculation considering a Mg2+ ion in gas phase indicates a larger binding energy for H2O (BEc = 326.5 kJ mol−1, MP2/aug-cc-pVTZ level) than for CO2 (247.7 kJ mol−1). Moreover, Mg2+ interaction with CO2 mediated through water would be characterized by a decidedly lower binding energy (80 kJ mol−1) at the same level of calculation. Strong and specific chemical interactions are then needed in order to increase the selectivity of the materials and their stability in operational conditions. Nevertheless, effective systems for CO2 removal must combine high selectivity and capacity with minimal energetic input to liberate the captured CO2 that means low interaction energies. In fact, the energy consumed per cycle is directly related to the sorption heat, if efficient heat recovery systems are not present. Stability in presence of moisture of the materials themselves is also an issue for example for metal–organic frameworks (see Section 4.3). Besides water, more corrosive contaminants as H2S are often present bringing to a further lowering of the cyclability of the separation system.
Summarizing, the ideal material for carbon dioxide capture from real flue gases and from atmosphere requires low synthetic cost, high stability, low heat of adsorption, long-term adsorption cycling, stability to contaminants and oxidative and high selectivity in presence of water. This means that on one hand the material need to be stable in presence of moisture and that the competition between CO2 and H2O is minimized.74
Separation technologies can be distinguished in four groups differing in the way the regeneration is performed: (i) PSA (pressure swing adsorption), (ii) VSA (vacuum swing adsorption), (iii) TSA (temperature swing adsorption) and (iv) ESA (electric swing adsorption). Other systems that have been reported can be seen as a combination of those processes. PSA and VSA differ on the pressures adopted. For PSA, the separation is conducted at about 5–6 bar, whereas the regeneration is performed by flushing the column with an inert gas at lower pressure (about 1 bar). For VSA, the pressures involved are lower, being the intlet at 1–1.5 bar and the outlet 0.1 bar. In TSA, the column is also heated during the regeneration by passing a hot gas through the column.75 For ESA, the desorption is achieved by heating the fixed bed using electricity,75 and it is then more effective than TSA if the adsorbent itself is a good electric conductor (e.g. graphitic carbons). Among those processes, TSA is particularly promising, owing to difficulties with compressing or applying a vacuum to large volumes of a low pressure flue gas stream, that would be required for pressure-swing or vacuum-swing adsorption.76
At this point, it is important to stress that on the applicative point of view, volumetric CO2 densities reached in a material are more significant than CO2 gravimetric densities.77 Nevertheless, in general these data are not available in the articles. For this reason, in the following only the gravimetric densities are reported in order to allow the comparison with the different materials. It would be useful that in future publications, the volumetric capacities will be reported along the gravimetric ones.
Another important consideration derives by reminding that three factors are at the basis of any separation process: (i) thermodynamics; (ii) molecular sieving and (iii) kinetic effects. For reason, separation properties of materials would be accurately characterized only in studies performed in dynamic conditions. Nevertheless, those measurements require the use of reactors, mass spectrometers and large quantities of sample, making them trickier measurements than static volumetric measurements. For these reasons, in most part of the articles only the selectivities derived from static measurements are reported (adsorption selectivity, Sads).76 In the following, selectivity will be then often synonym of adsorption selectivity. Nevertheless, I am aware that the permeation selectivities Sper are the correct parameter to be considered, defined as the product of the adsorption (Sads) and the diffusion (Sdiff) selectivities.78 Sads are almost coincident with Sper only when high correlation effects in molecular jumps from one adsorbed site to another are present for the different species (Sdiff = 1).78 This is true for example in materials characterized by 1D channel pores or by small pore openings.78 If the correlation effects are negligible (Sdiff being the quotient of the Maxwell–Stefan diffusivities of the pure components in the material), then Sads represents only an approximation of Sper. The relative scale of material performances can be then different in dynamic conditions. In fact, higher Sads is not directly correlated with high Sdiff being the former enhanced by high energetic of interactions whereas the second decreases, if high correlations are not present. Diffusion has to be enhanced (by adopting higher operational temperatures, larger intercorrelation and pore openings) if a large difference in the adsorption strength of the two gas components is present.
Computer simulations aimed to predict selectivities of materials have shown to be a particularly accurate and easier tool in some cases with respect to the experimental counterpart. Nevertheless, in this case the use of rigid framework has to be carefully avoided, constituting a higher approximation with respect to more negligible factors as dimensions of the particles and defects.79 This is particularly true in the case of metal organic frameworks, materials that can be characterized by drastic structural changes upon adsorption.80,81
3. The Mg2+ ion
Besides few exceptions, magnesium is in general in its ionic form Mg2+. The ions reactivity or, more generally, the ions ability to polarize adsorbate can be predicted through their charge/radius ratio but also through their electrostatic effects on the chemical properties of ligands to which they are coordinated. A fairly clear concept of the magnitude of this effect can be acquired by considering the acid strengthening effect of a divalent metal ion on a coordinated water molecule.82 In Table 4, the pKa for water coordinated to different divalent ions is reported. Ionic radii for the same ions in tetra- (rtetr) and hexacoordinate complexes (rhexa) are also presented.82 If the pKa values are compared with that of pure water (15.7), it is evident that all of them are lower than this value and in most cases the smaller rhexa, the lower the pKa, i.e. larger the ion reactivity. For water molecules coordinated to ions in four coordination, an even lower pKa is then expected for all the ions. It is noteworthy that, if the Be2+ ion is excluded, Mg2+ has a pKa value very close to the median of the values reported in Table 4. This suggests as catalysts involving Mg2+ in the active site would have a good reactivity but also a relatively low stabilization of the reaction products. For what concerns separation processes, a lower regeneration energy of the Mg-based systems with respect to other M2+-based systems can be predicted on these basis.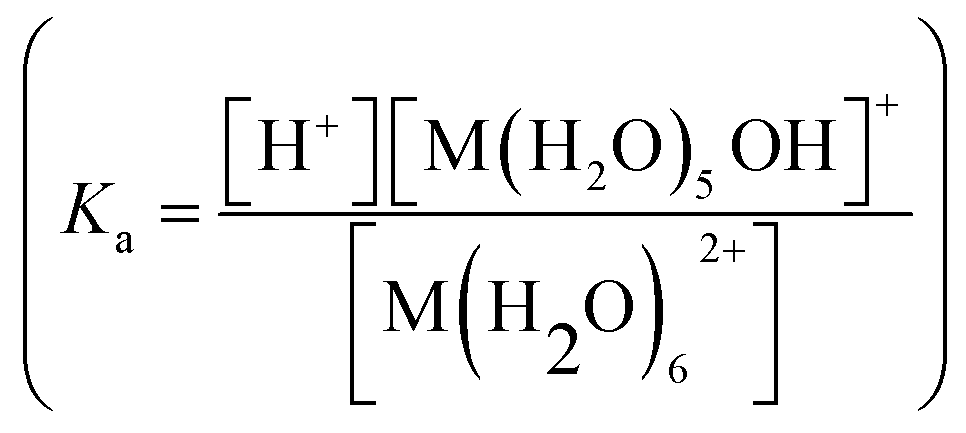 for water hexacoordinated to divalent M2+ ions and the ions radius in hexacoordinate (rhexa) and tetracoordinate complexes (rtetr), as from XRD measurements. Data from ref. 82 and references therein
for water hexacoordinated to divalent M2+ ions and the ions radius in hexacoordinate (rhexa) and tetracoordinate complexes (rtetr), as from XRD measurements. Data from ref. 82 and references therein
| pKa | rhexa (Å) | rtetr (Å) | |
|---|---|---|---|
| Ba2+ | 13.1 | 1.49 | — |
| Be2+ | 4.3 | 0.59 | 0.41 |
| Ca2+ | 12.5 | 1.14 | — |
| Cd2+ | 9.8 | 1.09 | 0.92 |
| Co2+ | 9.4 | 0.89 | 0.72 |
| Fe2+ | 8.4 | 0.92 | 0.77 |
| Mg2+ | 11.4 | 0.86 | 0.63 |
| Mn2+ | 10.1 | 0.97 | 0.8 |
| Ni2+ | 9 | 0.83 | 0.69 |
| Zn2+ | 9.6 | 0.88 | 0.74 |
Nevertheless, CO2 is a quadrupolar molecule and, for this reason, it would be more sensitive to the polarity of the bond involving Mg2+ than to the polarizing ability of the ion alone when Mg2+ is part of a material. In this case the difference in electronegativity between the ions involved in the bond is a more precise indication of the polarity of the materials surfaces.61 The large electronegativity difference between Mg and C, N and O (the most common Mg2+ ligands in materials) allows to predict a high polarity in the bond involving Mg and then a high affinity towards CO2. This has been actually verified in comparative studies on oxides61 and metal–organic frameworks.83
4. Mg-based materials involved in the CO2 cycles
4.1. Enzymes
Mg2+ is present as essential cofactor in many enzymatic reactions going from methylation, phosphorylation and carboxylation.15,82 Its large presence is likely related to its large abundance in nature but also to its small ionic radius (see Table 4), making it an essential factor in electrophilic catalysts.82 In enzymatic reactions, in fact, the electrostatic effect of the metal on the chemical properties of the ligands to which it is coordinated82 plays a crucial role firstly in determining the acidity of the coordinated water. Carboxylases are enzymes that allows carboxylation, that is carbon dioxide fixation. In general, carbon dioxide is present as bicarbonate (HCO3−) in cells.82 Nevertheless, bicarbonate is unreactive as a carboxylating agent, whereas carbon dioxide is electrophilically reactive.82 For this reason, the first step in the carboxylation reactions is often constituted by the conversion of bicarbonate to carbon dioxide through the mediation of adenosine triphosphate (ATP).Among carboxylases, six of them can present Mg2+ as metal cofactor in the active site:15 Rubisco, PEPC, PEP carboxykinase, acetyl-CoA carboxylase, proprionyl-CoA carboxylase and pyruvate carboxylase.15 In particular, Rubisco and PEPC are the two most important carboxylases enzymes. They differ in the form in which is CO2 at the beginning of the reaction: Rubisco uses dissolved CO2, whereas the one used by PEPC is originating from the dehydration of bicarbonate.
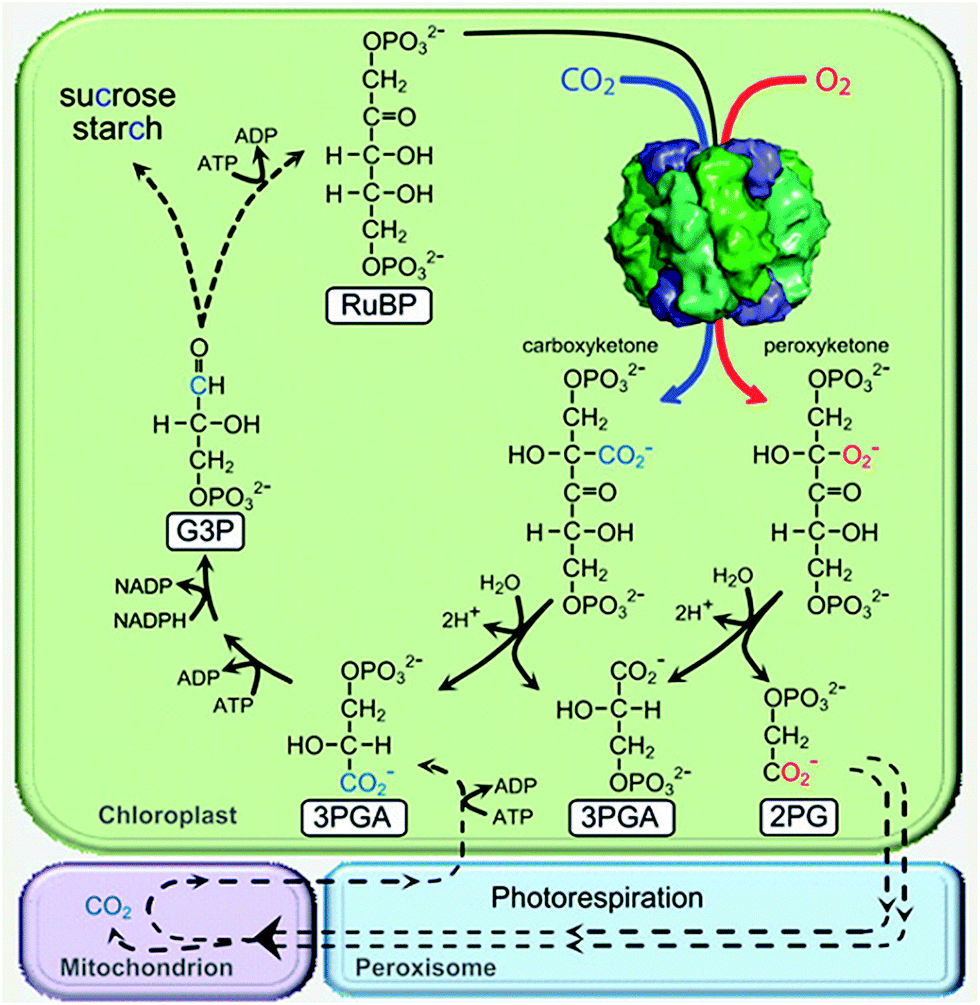 | ||
| Scheme 3 Simplified scheme illustrating how CO2 fixed to RuBP by Rubisco is distributed among the resulting two molecules of 3PGA that feed into the photosynthetic Calvin cycle to produce triose phosphates (glyceraldehyde 3-phosphate [G3P]) for carbohydrate synthesis or RuBP regeneration. The contrasting oxygenation reaction of Rubisco produces 2-phosphoglycolate (2PG), which requires the photorespiratory pathway to recycle it back to 3PGA. Photorespiration is a complex pathway that involves four subcellular compartments and multiple enzymatic steps (represented by dashed lines), requires additional energy (ATP), and results in a loss of fixed CO2 in the mitochondria. Reproduced with permission from ref. 84. Copyright 2011 American Society of Plant Biologists. | ||
The structure of Rubisco as in Spinacia olearia is described in Fig. 5a and b while in part c only the 8 pockets and corresponding active sites of the enzyme are reported. In part d, a scheme of the active site is reported, with bound to the Mg2+ ion and CABP, an isomer of the reaction transition state. An atomistic representation of part d, aimed to have a closer view on the Mg2+ coordination, is reported in parts e and f of the same figure. In plants and cyanobacteria,88 this enzyme has a mass of about 550 kDa and it is constituted by eight large (L) and eight small (S) subunits, with each couple differentiated in Fig. 5 by means of different colors. Each of the eight L subunits hosts an active site (Fig. 5c) situated on the external surface of the enzyme. Among all the species present in the pockets, only four residues and the Mg2+ ion are responsible of the enzyme activity.
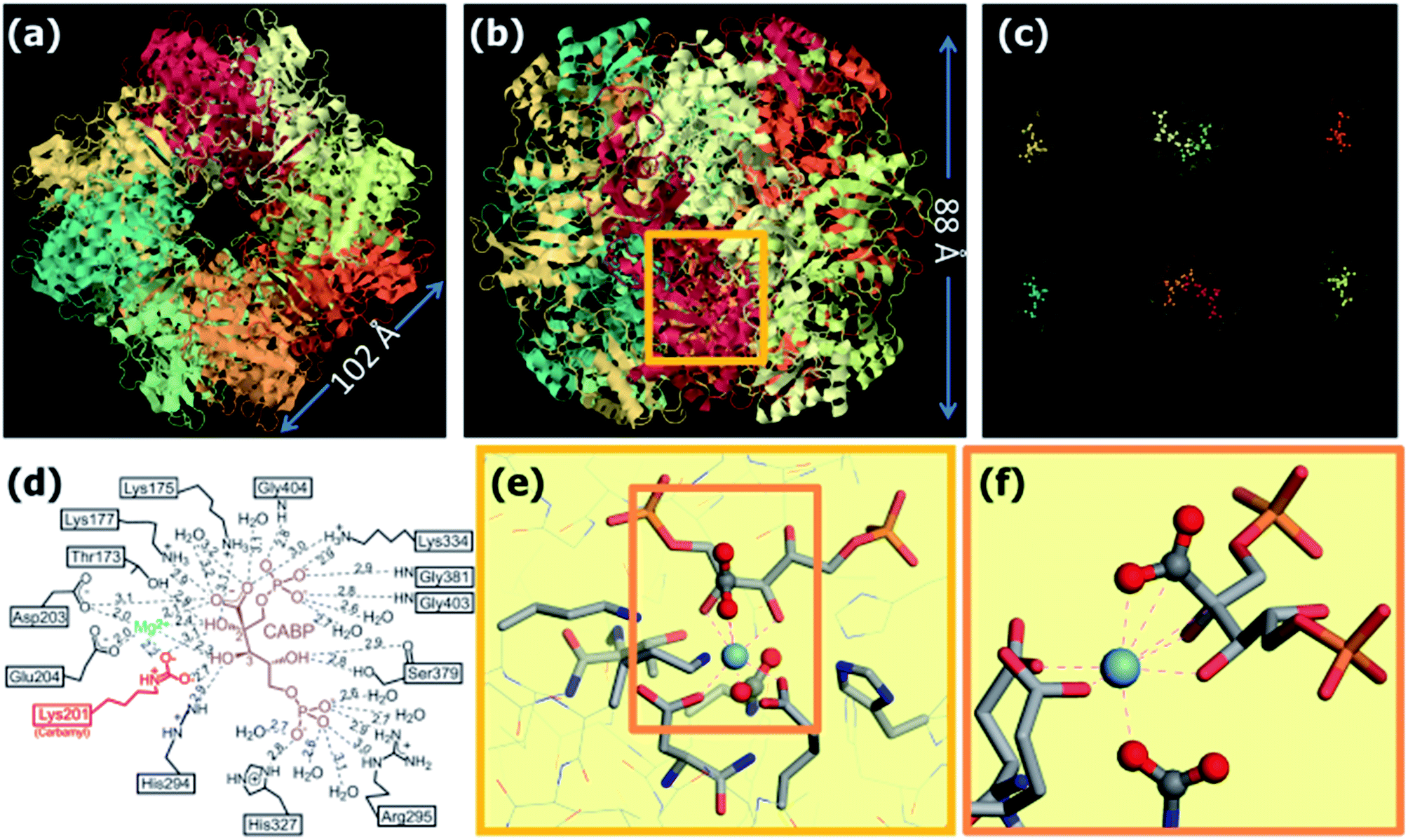 | ||
| Fig. 5 Rubisco with Mg2+ and the transition state analog CABP bound to the active site from Spinacia oleracea (Protein Data Bank ID: 8RUC, resolution of 1.6 Å)88 shown from two perspectives: (a) looking down the fourfold axis of symmetry and (b) rotated 90°. The eight subunit couples L–S (large and small) are represented with different colors (images obtained by the JMol software).100 (c) The eight pockets (one per L unit) with the ligands are represented using the same orientation as in part (b). (d) Schematic 2D representation of the active site complex, showing Mg2+, CABP, and all of the active site contacts, including the carbamyl-Lys201, with interatomic contact distances given in angstroms as in the 1RXO structure (PDB ID). Adapted from ref. 82, Fig. 8.23D from p. 423. By permission of Oxford University Press, USA. (e and f) Atomistic view of the coordination around the Mg2+ ion in two different orientations and magnifications. The CO2 units (that is the one directly involved in the carboxylation reaction and the one incorporated in the carbamyl unit of lysine) are represented as ball and stick atoms. The atoms at a distance lower than 3 Å from Mg2+ are linked to the ion through dashed lines. The atoms are represented according to following color code: oxygen (red), carbon (grey), sulfur (yellow), phosphor (orange), magnesium (light blue), nitrogen (dark blue). Hydrogen atoms, being absent in the determined XRD structure, were omitted. | ||
In the reaction catalyzed by Rubisco, a minimum of two carbon dioxide molecules are needed: one associated to the activation of the enzyme, while the second one participating to the carboxylation reaction. The activation of the enzyme would proceed through a two step mechanism: in the first step, one CO2 molecule reacts with the ε-amino group of a lysine residue101 (Lys201 in spinach enzyme) causing its carbamylation. In this part, Mg2+ would assume a rare pseudotetrahedral coordination, with three water molecules and the forth position occupied by CO2. During the second step, Mg2+ is then bound to the enzyme, through the N-carbamyl group formed in most part of the complexes.101 Mg2+ is in an octahedral coordination at this point, coordinating a water molecule that plays an important role in the whole reaction. Since this step, Mg2+ would maintain an octahedral coordination, although differently distorted.86 The carboxylation and cleavage of RuBP then take places through a six step reaction (extensively discussed in the literature, see in particular ref. 86): (i) coordination of RuBP; (ii) coordination of CO2; (iii) carboxylation of RuBP; (iv) hydration; (v) RuBP cleavage to a 3-PGA molecule and a carbanion intermediate and (vi) protonation of the carbanion with the formation of a second 3-PGA molecule. Recent calculations indicate that the coordination and activation of CO2 would happen through an acid base mechanism given by a concerted action of the RuBP and the water molecule coordinated to Mg2+.86,102 The Gibbs free energy of CO2 coordination (solvated system) in the step (i) has been calculated to be of 21 kJ mol−1 by means of quantum mechanics calculations (PBE-D).102 The structure around Mg2+ in the transition state that precedes the RuBP cleavage (step v) is reported in Fig. 5d–f. This transition state is estimated to be about 100 kJ mol−1 higher in energy with respect to step (i). In these figures, it is evident that Mg2+ is in a distorted octahedral coordination. Each vertex of the octahedron is occupied by an oxygen atom, where the two apical positions are occupied by one oxygen of the two pristine CO2 molecules. The other four oxygens are due to two O of the CABP molecule and the remaining two are belonging to two carboxylate groups (Asp204, Glu203 residues).82 All these oxygens are at distances between 2.3 and 2.4 Å from Mg2+. It is interesting to notice that the CO2 molecule is strongly bent (∠O–C–O = 118°) and that both the C and one O are at a distance lower than 3 Å from Mg2+.88 The importance of Mg2+ in the reaction is evidenced by the fact that removing it or exchanging it with a Ca2+ ion, the enzyme looses completely its catalytic activity.82 This is likely related to the importance of the coordinated water in the reaction: Mg2+ has the ability to polarize water more than Ca2+ (see Table 4) without having the ability to favor its reaction with CO2, as for example a Zn2+ ion would do (the active site in the carbonic anhydrase enzyme).
All known PEPC are tetrameric enzymes: a PEPC can be described as a dimer of dimers with respect to the subunit constant (see Fig. 6a),110 having a molecular weight of about 440 kDa. PEPC activity is controlled in an allosteric manner, through the presence of activator or inhibitor molecules. In the activation step, PEPC binds firstly the metal cofactor, constituted by a divalent cation, either Co2+, Mn2+ or Mg2+.82,92,103,105 This metal center will be necessary firstly in PEP coordination but it plays also a leading role in the last part of the reaction process, coordinating contemporaneously the enolate intermediate and CO2 bringing to the formation of oxaloacetate. After coordination of the metal by the enzyme, PEP is bound through a bidentate coordination to the metal. In Fig. 6b, the PEPC structure around the metal center of the enzyme from Escherichia coli (Protein Data Bank ID: 1JQN, resolution of 2.35 Å)107 is reported. In this structure, PEPC is bound to Mn2+ (metal cofactor), to a PEP analog (3,3-dichloro-2-dihydroxyphosphinoylmethyl-2-propenoate, DCDP), and to aspartate (allosteric inhibitor). It is evident that the metal ion is in a roughly tetragonal bipyramidal coordination with all the vertex at an average distance of 2.1 Å. This distance is significantly shorter that the one observed in Rubisco structure. As for the Rubisco metal center, all the first neighbours are six oxygen atoms: two of them belonging to the PEP moiety, two to water molecules and two oxygens due to two different COO− units (that is the ones relative to Asp603 and Glu566 residues in the inactive form of Escherichia coli PEPC). This is schematically represented in step 2 of Fig. 6c. Then the carboxylation reaction proceeds through a three step mechanism (step 3–5 in Fig. 6c). (i) Firstly, bicarbonate anion is coordinated by three arginine units in the pocket110 and acts as a nucleophile to attack the phosphate group in PEP dissociating it in a carboxyphosphate and in the very reactive enolate form of pyruvate; (ii) hystidine (H177 in Escherichia coli) then modulates the proton transfer on the carboxyphosphate through its diazolate ring bringing to its dissociation in a phosphate group and in a CO2 molecule; (iii) the reaction of CO2 with enolate happens with the formation of oxaloacetate and the protonation of the phosphate group [PO4]2− to [PO3OH]−.103,107
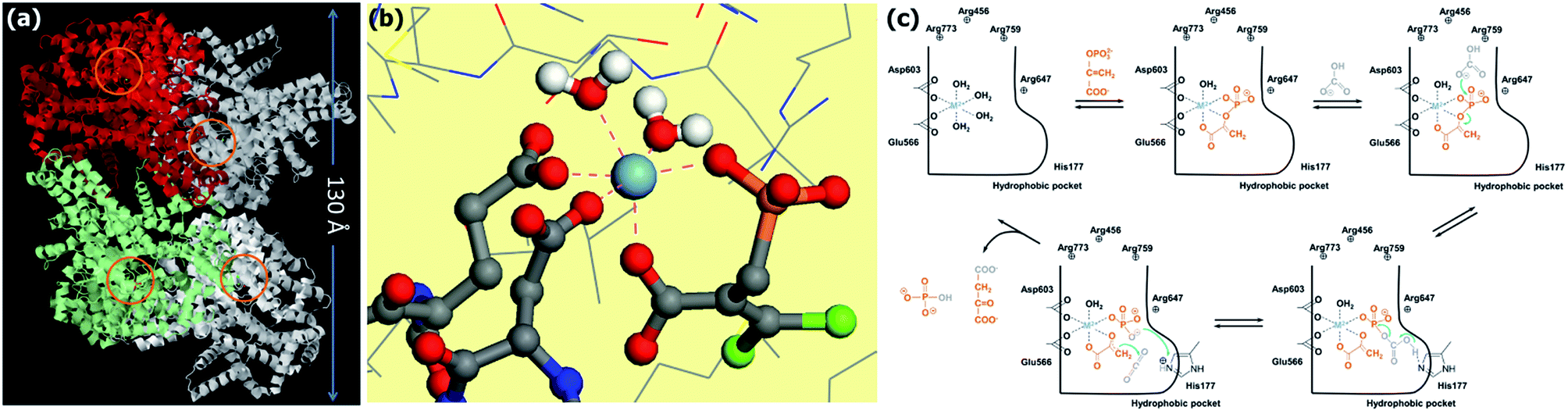 | ||
| Fig. 6 PEPC from Escherichia coli with Mn2+, phosphoenolpyruvate analog (3,3-dichloro-2-dihydroxyphosphinoylmethyl-2-propenoate, DCDP), and aspartate (allosteric inhibitor) bound to the active site (Protein Data Bank ID: 1JQN, resolution of 2.35 Å).107 (a) Ribbon diagram of PEPC view along the front C2 axis. The two subunits of one of the two dimers are differentiated by green and red colors (image obtained by the JMol software).100 The position of the four active sites is indicated by orange circles. (b) Atomistic view of the coordination around the Mn2+ ion in interaction with DCDP. The atoms at a distance lower than 2.4 Å from Mn2+ are linked to the ion through dashed lines. The atoms are represented according to following color code: hydrogen (white), oxygen (red), carbon (grey), sulfur (yellow), phosphor (orange), manganese (light blue), nitrogen (dark blue), chlorine (green). Hydrogen atoms not belonging to water molecules are omitted. (c) PEPC enzymatic mechanism as in ref. 107 converting bicarbonate and PEP to oxaloacetate and phosphate. | ||
4.2. Grignard reagents
Grignard reagents are obtained from the reaction of an alkyl, alkenyl or aryl halide (RX, X = Cl, Br or I) with elemental magnesium:| RX + Mg → RMgX |
The preparation of a Grignard reagent requires in general an ether solvent (usually diethyl ether or tetrahydrofuran). The reagent is then solubilized through the coordination to Mg by ether oxygens (see Fig. 7a). Completely anhydrous conditions need to be adopted because of the high Grignard reagent reactivity with water. Also oxygen presence has to be carefully avoided. In ether solution, Grignard reagents are better represented through the Schlenk equilibrium mixture:
| 2RMgX R2Mg + MgX2 |
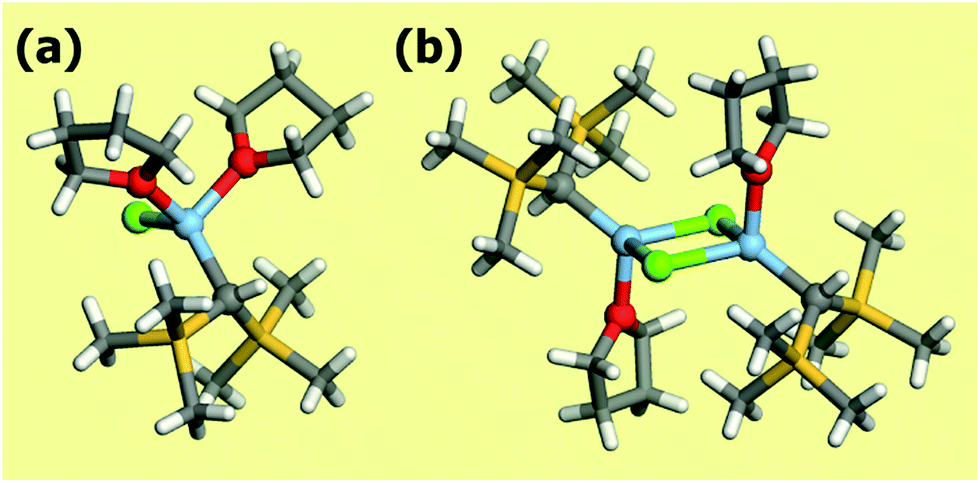 | ||
| Fig. 7 Mono- (a) and dimeric forms (b) of the Grignard reagent chloro-(bis(trimethylsilyl)methyl)-bis(tetrahydrofuran)-magnesium. Structures from ref. 117. The atoms are represented according to following color code: hydrogen (white), oxygen (red), carbon (grey), silicon (yellow), magnesium (light blue), chlorine (green). | ||
The strong separation in charge of C and Mg in Grignard reagents is expected to be particularly suitable to activate CO2. In fact, the reaction of these compounds with CO2 is a well known two step reaction and it is among the simplest reactions to form new C–C bonds. The synthesis is performed by using dry ice or bubbling CO2 in the solution:
| RMgX + CO2 (RCOO)⋯(MgX)+ |
| (RCOO)⋯(MgX)+ + HA RCOOH + MgXA |
This reaction allows to fix the whole carbon dioxide in an organic compound analogously as in Rubisco and PEPC enzymes by consumption of a reagent. Once formed, the newly affixed carbon of the carboxyl group can be modified to give a plethora of other substituents. Nevertheless, in ideal conditions a stoichiometric amount of Grignard reagents is consumed in the reaction. Life cycle assessments of this process would be particularly interesting to state if this reaction is actually having a positive carbon footprint or a negative one.
4.3. Metal organic frameworks
Metal organic frameworks (MOFs) are a relatively new class of crystalline materials which structure is constituted by the linking of metal oxide/metal clusters through organic linkers. At difference of many organometallic structures, in these materials a strong bond exists between the organic and the inorganic parts, allowing the retaining of the material architecture also upon solvent removal. The materials so obtained are in general highly porous and the possibility to easily tailor them toward specific applications has been often stressed in the literature on the subject.118,119 Their conceptual design has been compared to the Geomag toy (a toy construction system consisting primarily of metal spheres and short connecting sticks). In fact, several organic and inorganic parts (called secondary building units) with different coordination and functionalities can be in principle freely combined.120,121 The possibility to change their pore width, dimensionality of the structure and chemical compositions is relatively easier for this class of compounds with respect to anyone else. Their applications are spread from catalysis,122,123 gas adsorption and separation, fuel cells, proton conductivity, protein adsorption,74,124… The first report about a MOF structure date in 1971.119 Nevertheless, only after the report of the first structure able to retain its porosity upon solvent removal, a big exploit has been experienced by MOFs125 with the creation of more than 20000 different materials.119,124 Most of the guinness materials among crystalline ones belong to the MOF class: NU-110 (the largest surface area material, 7140 m2 g−1),126 IRMOF-74-XI (the largest pore aperture, 98 Å)124 and MOF-399 (the lowest density, 0.126 g cm3).127 However, there are still some open challenges due to their large synthetic cost and their low stability, both on the thermal and chemical point of view. For what concerns this last point, most of MOFs are highly sensitive to water, also at atmospheric concentrations, hindering their practical use.118,128,129 MOFs have been subjected to several studies for CO2 storage and separation in different pressure and temperature conditions.3,63,129,130 Their structure makes MOFs ideal subjects for combined experimental and theoretical studies, allowing to couple the ability to fast screening of the possible structures of computational techniques with a direct application of the indications obtained in experiments.17,72,78,131,132 By using this strategy, materials with superior performances with respect to any component of other materials classes have been reported for both CO2 separation and storage.131–133
Most part of MOFs materials are based on transition metals. The use of s-block metal centers to construct the metal oxide units in MOFs is comparatively rare.134 This fact is related to the predominance of ionic forces and absence of well defined metal-oxide secondary building units making more difficult the prediction of the coordination geometry and then the rational design of MOFs with respect to transition metal-based ones.134 Nevertheless, the high abundance of these elements, and they cheap and non toxic nature makes the exploratory synthesis of the corresponding MOFs a worthwhile endeavour.134 Moreover, the larger ionicity of the metal–oxygen bond for s-block material with respect to transition metal centers is expected to be beneficial on interaction with a quadrupolar molecule as CO2. Among the s-block metals, magnesium is the most commonly used.134 The Mg-based MOFs tested as CO2 adsorbents/separators are reported in Table 1. A comprehensive list of the Mg-based MOFs reported in the literature so far is present in the ESI (Table S1†). It is important to notice that the most part of them has been reported only after the synthesis in 2005 of the first Mg-based MOF with permanent porosity.135
The relative abundance of Mg-MOFs is due to the structural chemistry of Mg2+, that is very close to the that of Zn2+, one of the most common metal in MOFs.134 Nevertheless, the lower atomic mass of Mg2+ with respect to transition metals confers to the Mg analogues higher surface areas and lower densities with expected larger performances on the gravimetric gas densities achievable in storage and separation. Volumetric gas densities (a more important parameter in CO2 separation, see Section 2.3)77 are instead expected to be lower. Another important point that hinders the practical use of Mg-based MOFs is related to their low stability in presence of water due to the large ionic character of Mg–O bonds.128 Mg2+ has a high affinity for oxygen donor atoms of water and other polar solvents, that can represent competitive ligands with respect to the organic linkers, facilitating material degradation.136 Although some Mg-MOFs have been claimed to be water resistant,137 a standardize method to evaluate the water resistance parameter is actually lacking and at the basis of contradictory observations reported in literature,129,138 likely related to different operation conditions. For what concerns catalytic reaction involving CO2, they are quite rare in MOFs122 and they are completely absent for Mg-MOFs materials.123
In all the structures reported for Mg-MOFs, Mg2+ is six-coordinated (see Tables 5 and S1†). Nevertheless, in most of them at least one of the ligands is represented by a solvent molecule (e.g. H2O, DMF) that can be in principle removed upon activation, that is upon thermal treatment of the material in vacuum or in inert gas flow. If the thermal stability of the structure is higher than the temperature needed to remove the solvent, at the end Mg2+ would show a coordination vacancy that represents a high energetic site for molecule adsorption. The possibility to create open metal sites in MOFs structure by activation has been reported in several cases.3,83,119,128,132,135,139–142 In this regard, particularly interesting are the isomorphous materials [M2(dobdc)(H2O)2] with H4dobdc = 2,5-dihydroxytereftalic acid and M = Ni, Co, Zn, Mg, Mn or Fe.3,83,141 These materials have been reported in literature with several acronyms, as CPO-27-M, M-MOF-74, IRMOF-74-I or M/DOBDC. Their structure is an honeycomb-like with the vertex of the hexagon occupied by filars of M–O units linked between them by dobdc units (see Fig. 8a). The metal center is in an octahedral coordination in the as-synthetized material, with one of the positions occupied by a water molecule. This molecule is removable upon degassing at 120 °C, so leaving open the metal center.139,143 Among the possible metal centers, M = Ni, Co, Zn, Mg have been compared for their CO2 adsorption performances.83 MOFs represent ideal systems to systematically study the roles of different open metal sites or functional groups on materials properties. The isotherms obtained in ref. 83 at 23 °C are reported in Fig. 9b (see also Table 5). It is evident that the CO2 uptake of IRMOF-74-I-Mg is twice larger than in any other member of the series.3,11,83 This is true in the whole 0–1 bar range, with significant uptakes also at pressures close to 0.39 mbar (0.22 mmol g−1).83
| Materiala | CNb | Framework dimensionality | SBETc (m2 g−1) | SLangmuirc (m2 g−1) | Vporc (cm3 g−1) | nCO2 @P,Tf,g (mmol g−1) | qiso (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a NH2-BDC = 2-amino-1,4-benzenedicarboxylate; DMF = N,N-dimethylformamide, H2bdc = 1,4-benzenedicarboxylic acid, DMA = dimethylacetamide, H2bpdc = 4,4-biphenyldicarboxylic acid, H3btc = 1,3,5-tricarboxylic acid, H2-3,5-PDC = 3,5-pyridine dicarboxylic acid, H2-2,4-PDC = 2,4-pyridine dicarboxylic acid, H3-3,5-PZDC = pyrazole-pyridine dicarboxylic acid; H2NDA = 1,4-naphthalenedicarboxylic acid, phen = 1,10-phenantroline, H2hfpb = (hexafluoroisopropylidene)bis(benzoic acid); def = N,N-diethylformamide; ndc = 2,6-naphthalenedicarboxylate; D-H2Cam = D-camphoric acid; DMA = dimethylacetamide; H2tart = tartaric acid, dif = N,N-diisopropylformamide; H2OBA = 4,49-oxybisbenzoic acid; H4dobdc= 2,5-dihydroxy-1,4-benzene-dicarboxylic acid; BPTC = 2,2′,6,6′-tetracarboxybiphenyl; Boc =tertbutyloxycarbonyl; H3idc = 4,5-imidazoledicarboxylic acid; H4TTTP = 2′,3′,5′,6′-tetramethyl-[1,1′:4′,1′′-terphenyl]-4,4′′-dicarboxylic acid; formamidinium cation [Fmd+, (NH2–CH+–NH2)]; guanidinium cations [Gua+,C+(NH2)3]; TCPBDA2− = N,N,N′,N′-tetrakis(4-carboxyphenyl)-biphenyl-4,4′-diamine, ED = etilendiamine; TEPA = tetraethylenepentamine; mmen = N,N′-dimethylethylenediamine.b In parenthesis the number and the nature of solvent molecules coordinated to the most exposed Mg2+ species are reported.c Surface area and pore volume as obtained from N2 adsorption, if not otherwise specified.d Calculated value upon manual removal of the solvent molecules from the solvated structure.e Ar isotherm and pressure range adopted in the BET area calculation different from the standard one.f The technique used for the measurement is indicated in parenthesis: vol = volumetry (static conditions), ms = mass spectroscopy, tga = thermal gravimetrical analysis.g Conversion from original data in wt% made on the assumption that wt% = CO2 weight/(CO2 weight + sample weight) × 100, if not otherwise specified. | ||||||||
| {[Mg(Hidc)(H2O)2]·1.5H2O}n | 6(2H2O) | 1D | Low | Low | 0.06 | 0.27@195 K, 0.93 bar (vol) | — | 157 |
| {[Mg3(idc)2(H2O)5]·2H2O}n | 6(3H2O) | 3D | Low | Low | 0.02 | 0.31@195 K, 0.93 bar (vol) | — | 157 |
| MgTTTP (PCN-72) | 6(1DMSO) | 3D | Low | Low | Low | 3.30@195 K, 1 bar | — | 137 |
| 1.56@273 K, 1 bar | ||||||||
| 1.34@295 K, 1 bar | ||||||||
| [Mg2(HCO2)2(NH2-BDC)-(DMF)2]n | 6(1DMF) | 2D | — | — | — | 3.99@273 K, 1 bar (vol) | — | 136 |
| β-Mg(HCOO)2·2H2O | 6(2H2O) | 3D | — | — | — | 0.30@298 K, 0.93 bar (vol)g | — | 158 |
| α-[Mg3(O2CH)6] | 6(0) | 3D | 150 | — | 0.11 | 1.69@298 K, 1 bar (vol) | — | 159 |
| γ-[Mg3(O2CH)6] | 6(0) | 3D | 120 | — | 24.7 vol%d | 2.01@298 K, 1 bar (vol) | — | 160 |
| Mg(HCOO)3⊃(CH3)2NH | — | — | — | — | — | 0.30@298 K, 1 bar | — | 158 |
| [(Fmd)Mg(HCOO)3] | 6(0) | 3D | Low | Low | Low | 0.02@195 K, 1 bar | — | 161 |
| [(Gua)Mg(HCOO)3] | 6(0) | 3D | Low | Low | Low | 0.01@195 K, 1 bar | — | 161 |
| {[Mg2(H-3,5-PZDC)2(H2O)4]·H2O}n | 6(2H2O) | 2D | Low | Low | Low | 0.36@298 K, 1 bar (vol) | — | 123 |
| [Mg(3,5-PDC)(H2O)] (Mg-MOF-1) | 6(1H2O) | 3D | — | — | 38.7 vol% | 0.63@298 K, 1.01 bar (tga/over N2) | — | 162 |
| Mg2(dobdc) CPO-27-Mg, MOF-74-Mg, IRMOF-74-Ie | 6(1H2O) | 3D | 877 (ref. 163) | 1030 (ref. 163) | 0.37 (ref. 163) | 5.36@298 K, 0.1 atm (ref. 83) | 47 (ref. 83) | 77, 83, 124, 141, 144, 151 and 163 |
| 1495 (ref. 83) | 1905 (ref. 83) | 0.60 (ref. 124) | 8.0@298 K, 1 atm (ref. 83) | 47 (−ΔHads, VTIR)144 | ||||
| 1350 (ref. 124) | 1600 (ref. 124) | 0.21 (ref. 151) | 14.32@298 K, 33 bar (vol)141 | 38–43 (ref. 141) | ||||
| 1542 (ref. 141) | 2060 (ref. 164) | 15.66@278 K, 36 bar (ref. 141) | 42 (ref. 77) | |||||
| 1800 (ref. 164) | 886 (ref. 151) | 9.04@313 K, 40 bar (ref. 164) | ||||||
| 780 (ref. 151) | 6.70@473 K, 40 bar (vol)141 | |||||||
| 2.67@333 K in 15% CO2/N2 (flow-ms)151 | ||||||||
| Mg2(dobpdc), IRMOF-74-IIb | 6(1H2O) | 3D | 2510e,124 | 2940e,124 | 1.04e,124 | 0.17@298 K, 0.2 mbar (ref. 147) | 44 (ref. 147) | 124 |
| 1.16 (ref. 147) | 6.6@298 K, 1 bar (ref. 147) | |||||||
| IRMOF-74-III-CH3 | 6(1H2O) | 3D | 2640 | 3940 | 1.37 | 2.95@298 K, 1.07 bar | — | 118 |
| IRMOF-74-III-NH2 | 6(1H2O) | 3D | 2720 | 4130 | 1.44 | 3.17@298 K, 1.07 bar | — | 118 |
| IRMOF-74-III-CH2NHBoc | 6(1H2O) | 3D | 2170 | 2720 | 0.95 | 2.08@298 K, 1.07 bar | — | 118 |
| IRMOF-74-III-CH2NH2 | 6(1H2O) | 3D | 2310 | 3270 | 1.14 | 3.27@298 K, 1.07 bar | Reversible @120 °C | 118 |
| IRMOF-74-III-CH2NMeBoc | 6(1H2O) | 3D | 2220 | 2540 | 0.89 | 1.90@298 K, 1.07 bar | — | 118 |
| IRMOF-74-III-CH2NHMe | 6(1H2O) | 3D | 2250 | 3150 | 1.13 | 2.85@298 K, 1.07 bar | Reversible @120 °C | 118 |
| IRMOF-74-I-ED | 6(1ED) | 3D | — | 469 | 0.40 | 1.51@298 K, 400 ppm CO2 in Ar (flow-tga) | Reversible at 110 °C | 148 |
| IRMOF-74-I-TEPA-30 | 6(1TEPA) | 3D | 312.63 | 410.68 | 0.19 | 4.49@333 K in 15% CO2/N2 (flow-ms) | — | 151 |
| IRMOF-74-I-TEPA-40 | 6(1TEPA) | 3D | 132.24 | 230.5 | 0.15 | 6.06@333 K in 15% CO2/N2 (flow-ms) | — | 151 |
| IRMOF-74-I-TEPA-50 | 6(1TEPA) | 3D | 23.54 | 54.21 | 0.05 | 3.48@333 K in 15% CO2/N2 (flow-ms) | — | 151 |
| Mg(dobpdc)2-mmen | 6(1mmen) | 3D | 70 (ref. 10) | — | 0.02 (ref. 147) | 1.8@298 K, 0.3 mbar (vol)147 | 71 (ref. 147) | 10 and 147 |
| 3.9@298 K, 1 bar (vol)147 | ||||||||
| 3.14@313 K, 0.15 bar (vol)147 | ||||||||
| 1.05@298 K, in 1 h in Ar flow with 390 ppm CO2 (flow-ms)147 | ||||||||
| 2.52@313 K, in 15’ in 15% CO2/N2 (flow-ms)147 | ||||||||
| [Mg(TCPBDA)(H2O)2]·6DMF·6H2O (SNU-25) | 6(2H2O) | 3D | Low | Low | Low | 1.49@298 K, 1.01 bar | — | 150 |
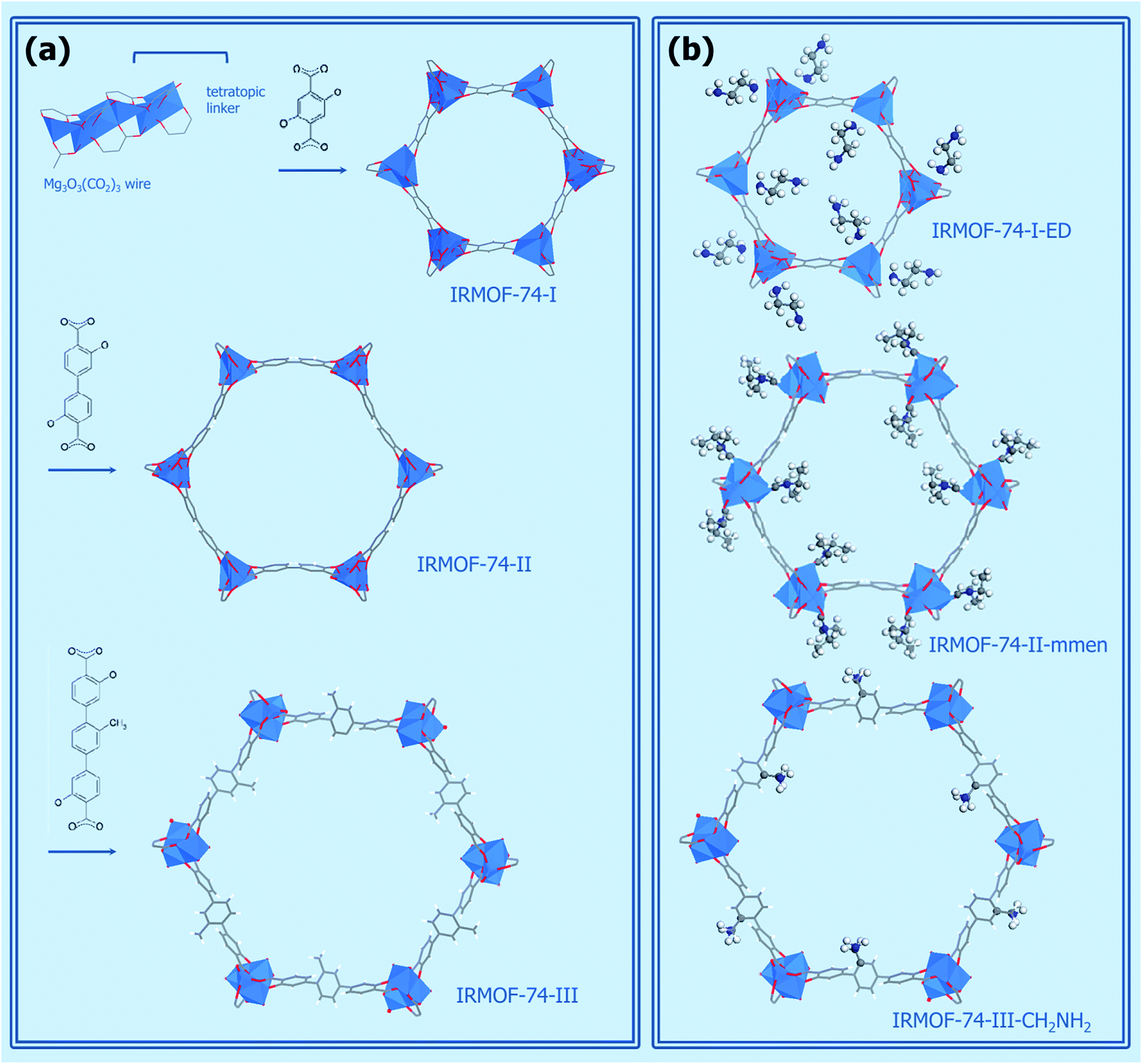 | ||
| Fig. 8 (a) Structures of the isomorphous IRMOF-74 materials for increasing dimension of the linker (from top to bottom, IRMOF-74-I, II and III) as from ref. 118, 120 and 147. (b) Exemplificative structures of amino-functionalised IRMOF-74 reported in literature.118,147,148 The atoms are represented according to following color code: hydrogen (white), oxygen (red), carbon (grey), magnesium (light blue), nitrogen (dark blue). Please notice that the structure reported in the top of part b as in ref. 148 is likely biased by symmetric constraints in the calculations. | ||
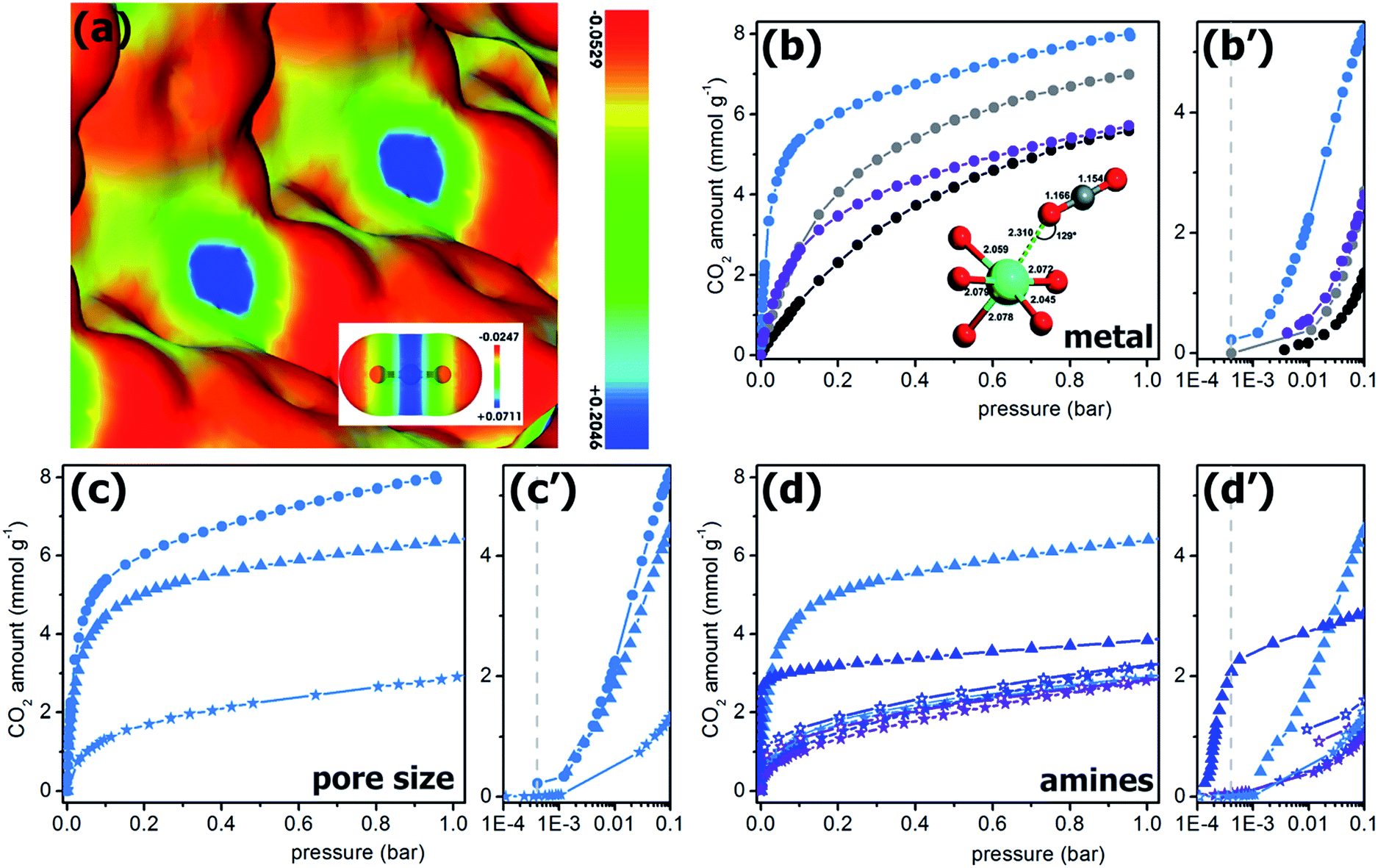 | ||
| Fig. 9 (a) Detail of the electrostatic potential of IRMOF-74-I mapped on the electron charge density isosurface (0.003 au). Red, green, and blue represent negative, zero, and positive values of the potential (a.u.), respectively. In the inset the electrostatic potential map obtained for CO2 is reported. Adapted from ref. 144. Reproduced with permission. Copyright 2010 American Chemical Society. (b–d) CO2 adsorption isotherms obtained at 25 °C on IRMOF-74 materials. The three plots compare the curves obtained for materials differing in (b) the metal center, (c) the dimension of the linker or (d) in the presence and nature of aliphatic amines groups. Full and empty scatters refer to the adsorption and desorption branch of the isotherm, respectively. Plots obtained by digitalization of the data reported in ref. 83, 118 and 147 in order to allow a direct comparison. The scatters refer to the points of the original curves used for the digitalization. The legend is: (b) IRMOF-74-I-Mg (light blue scatters), Co (grey), Zn (black) and Ni (violet); (c) IRMOF-74-I (circles), IRMOF-74-IIb (triangles) and IRMOF-74-III (stars); (d) IRMOF-74-IIb (light blue triangles), IRMOF-74-IIb-mmen (dark blue triangles), IRMOF-74-III (light blue stars), IRMOF-74-III-CH2-NH2 (dark blue stars) and IRMOF-74-III-CH2-NHMe (violet stars). (b′–d′) Inset in the 0–0.1 bar pressure region. The vertical dashed line marks the value of current partial pressure of CO2 in the atmosphere (0.4 mbar).2 The cluster reported in part (b) reports the geometry of the CO2 adsorption on the metal site in IRMOF-74-I-Mg as obtained from periodic calculations (B3LYP-D*/TZVp). Reproduced with permission from ref. 144. Copyright 2010 American Chemical Society. The atoms are represented according to following color code: oxygen (red), carbon (grey), magnesium (light blue). | ||
Although characterized by a larger surface area, the exceptional CO2 uptake of IRMOF-74-I-Mg is more related to the higher adsorption energy measured in this material with respect to the others of the series (47 versus 41 and 37 kJ mol−1 for Ni and Co analogues, respectively).83 This may be attributed to the larger ionic character of the Mg–O bond.83 A detail of the electrostatic potential map in the pores of this MOF as obtained at the B3LYP-D*/TZVp level is reported in Fig. 9a. The high ionicity of this structure in the region around the metal site is well evident. This map shows in fact that there is a strong positive potential (blue regions) on Mg2+ site and a strong negative potential around the oxygen belonging to the carboxylate group of the organic ligand (red regions).144 Quantum mechanical calculations indicated that Mg2+⋯OCO complex would not possess a linear but an angular geometry in this material in order to maximize the lateral interaction between the CO2 molecule and the oxygen atom of the carboxylic group Ocarbox (C⋯Ocarbox distance 2.975 Å, see Fig. 9b).144 The calculations indicated a remarkable role played by the dispersion contribution, which roughly accounts for one-half of the total gas–solid interaction energy.144 A similar finding was reported in a combined DFT/CC and IR spectroscopic study on carbon dioxide adsorption on the zeolite H-FER.145 Accordingly, also dynamic separation measurements performed in flow for CO2/CH4 and CO2/N2 indicated clearly higher performances of the Mg2+-based material with respect of the other IRMOF-74-I.138
Similar results were obtained considering MOFs where the cations are hosted as extra-framework species (rho-ZMOF).146 It was computationally predicted that the isosteric heat of adsorption at the lowest coverage increases with the charge-to-diameter ratio of the cation for CO2 adsorption and separation, being Mg2+ rho-ZMOF the best performing material after Al3+ rho-ZMOF. Moreover, because of its larger pore volume, Mg2+ rho-ZMOF would possess the highest total capacity of the whole series also in this case.
IRMOF-74-I-Mg represents the most investigated MOF for CO2 storage and separation.3,11,83,141,144,149 This is due to its superiority in performances in the p < 1 bar range not just with respect to its isomorphous materials, but also with respect to all the other MOFs. It is also characterized by a very high CO2/N2 selectivity (175 at 0.15 bar CO2, with a purity of CO2 in the adsorbed phase of 97%),76 although lower than CaA zeolite (250).76 Moreover, in this range its CO2 volumetric capacity is very close to the one reported for NaX,3,10 the reference material in CO2 separation studies (see Tables 5 and 7). To explore the effect of the pore size of the material on the CO2 capacities, IRMOF-74-I analogues have been synthesized by using larger linkers, exploiting the peculiar property of MOFs to allow easy tailoring and modification of the materials: the structure of IRMOF-74-I isomorphous having biphenyl (two isomorphs: IRMOF-74-IIa,124 and IRMOF-74-IIb,147) and triphenyl (IRMOF-74-III)118 linkers are reported in Fig. 8a. The corresponding CO2 isotherms obtained at 25 °C for pressure ≤ 1 bar are reported in Fig. 9c, where they are compared with the IRMOF-74-I one. Although the larger pore volume of IRMOF-74-IIb and IRMOF-74-III, their CO2 capacity is lower and decreases with the pore size. This effect was expected because of the low pressure considered and the importance of dispersion forces in CO2 interaction with material surfaces, also in presence of highly polarizing sites.17,144,145 The potential of two opposite pore walls is additional only up to a limit pore dimension. A glance to Fig. 9c seems to indicate that whereas for IRMOF-74-IIb (light blue triangles) such an effect is still present, in IRMOF-74-III (light blue stars) it is completely lost. In fact, this material stores only 2.95 mmol g−1 of CO2 at 25 °C and 1 bar, that is about one forth of IRMOF-74-I in the same conditions. The strong effect of pore dimension on CO2 adsorption has been observed also for frameworks which combine the properties of interpenetration with the presence of coordinatively unsaturated sites150 as Mg-N′-tetrakis-(4-carboxyphenyl)-biphenyl-4,4′-diamine).150
Although promising, IRMOF-74-I needs still some improvements: first of all, it has been proven to be difficult to regenerate, being only the 87% of its capacity recovered after flushing with CH4 at RT.138 Moreover, it has been theoretically predicted149 (and then experimentally verified)128,148 that it rapidly loses its CO2 capacity under humid conditions. Presence of other trace flue contaminants such as SO2 and SO3 fast poison open metal sites.10 Regeneration in presence of moisture causes the hydrolization of the material. Even more important for applications, its CO2 adsorption capacities degraded by long term storing the material under both dry or humid conditions.148
Properties of porous solid materials can be easily modified by impregnating or tethering active groups onto their surface.11 Analogously to silica-based materials, metal–organic frameworks properties have been improved in the CO2 sorption by alkyl amines grafting. Alkylamines are able to chemisorbs CO2, leading to higher adsorption enthalpies after grafting (see Table 5), facilitating CO2 adsorption in the lower pressure regimes (see Fig. 9d). The presence of water in this case is even beneficial on the adsorbed CO2 instead of hindering its chemisorption, because it can allow to double the CO2 uptake per metal site.11
Moreover, the presence of amines is expected to confer to the materials a higher stability towards water because in presence of open metal sites, amines are grafted on those sites hindering their accessibility to water and then the hydrolysis of the metal–oxygen bond.10 Also regenerability of the material/cyclability appears to be improved by their presence.148 Amine grafting in MOFs was reported to be possible either through post-synthetic modification of the framework130,147,148,151–154 (analogously to silica-based materials),155 or incorporating them during the synthesis by using an appropriate linker.118 In post synthesis methods, as stated above, the grafting happens through the bonding between the open metal site and the amine molecule. The high crystallinity of MOFs and the large separation in space between the metal sites, avoid overloading of the material and a more stable grafting on their surfaces than in silica materials, providing higher stability upon cycling.63 Between the two methods, the second approach makes intrinsically more stable the grafting of amines improving the cyclability of the materials.
The presence of aliphatic amines in the framework has been actually proven to improve the material stability under air and moisture,118,148 their regenerability and also their affinity toward CO2, allowing MOFs to reach a degree of affinity toward CO2 and stability close to amine-modified oxides.148 Both the grafting approaches have been applied to representatives of Mg-IRMOF-74 family: IRMOF-74-I, IRMOF-74-IIb and IRMOF-74-III. In particular, for IRMOF-74-I and IRMOF-74-IIb effective post-synthesis grafting procedures were reported allowing a high dispersion of ethylenediamine (ED)148 and tetraethylenepentamine151 (TEPA) in IRMOF-74-I and of N,N′-dimethylethylenediamine (mmem)147 in IRMOF-74-IIb (see Fig. 8b). On the contrary, in IRMOF-74-III different aliphatic amines have been introduced during the synthesis by using appropriate linkers: IRMOF-74-III-NH2, –CH2NHBoc, –CH2NMeBoc, –CH2NH2, and –CH2NHMe.118 It is evident from the comparison of part a and b of Fig. 8 as the introduction of an amine functionality strongly compromises the free pore volume of IRMOF-74-I and -II whereas IRMOF-74-III, because of its large pore dimension, would maintain a high porosity (see Table 5). Correspondingly, a strong decrease in the CO2 capacities at 1 bar for IRMOF-74-IIb-mmen with respect to IRMOF-74-IIb was observed (see Fig. 9d), whereas the pristine and the modified IRMOF-74-III show essentially the same capacity at 1 bar. Nevertheless, IRMOF-74-IIb-mmen adsorbs at least twice the CO2 amounts than the alkylamine-modified IRMOF-74-III. This is likely related to the nature of the alkylamine used. In fact, IRMOF-74-I-ED148 showed completely comparable CO2 adsorption than IRMOF-74-IIb-mmen for CO2 partial pressures close to atmospheric one (1.51 vs. 1.8 mmol g−1, respectively).147,148 IRMOF-74-I-TEPA-40 (ref. 151) showed higher CO2 capacities than any other MOFs for a flow containing 15% of CO2, doubling the value reported for IRMOF-74-IIb-mmen (see Table 5).147 In this study, Cao et al.151 evaluated also the influence of amine content on the adsorption capacity and they found as CO2 capacities have a bell-bottomed dependence with respect to the concentration of the amine, likely associated to diffusion problems at high amine content. It is interesting to compare the shape of the isotherms reported in Fig. 9d′ in the 0–0.1 bar pressure range. Only for alkylamine modified materials, the isotherms show a S-shaped behaviour that is particularly evident for IRMOF-74-IIb-mmen. Such a shape indicates that small drops in the CO2 pressure are expected to cause a fast release of the CO2 chemisorbed in the material and then an easy regeneration in pressure swing adsorption plants. The presence of amines is then beneficial on the CO2 capacity only in the 0–0.1 bar pressure regime, making the material more selective for CO2 capture from post-combustion flue gases (15% CO2) or even from atmosphere (400 ppm). This effect is less evident for modified IRMOF-74-III materials, that shows essentially the same capacity of the unmodified material in the whole pressure range. Nevertheless, the difference between their adsorption (full scatters) and desorption (empty scatters) branches clearly indicates the formation of carbamates due to the reaction between the amine and CO2 (see Fig. 9d′, violet and dark blue curves). Further, a larger stability of the separation performances in presence of water was observed for all the alkylamine modified materials. For example, the breakthrough time obtained for CO2/N2 separation in dry and moisture conditions was coincident for IRMOF-74-III-CH2-NH2, whereas the parent IRMOF-74-III-CH3 showed a 80% decreases if water was admitted in the gas flow.118 This stabilization effect is obviously not verified if aromatic amines are introduced in the framework: in fact, although their introduction causes an increase of the energetic of adsorption because of cooperative effects,17,156 they are not able to chemically react with CO2.
Although the higher sorption heat (71 vs. 44 kJ mol−1 for IRMOF-74-IIb with and without mmen, respectively)147 due to the formation of carbamate/bicarbonate species, lower temperature for the full regeneration of the material were reported the amine modified IRMOF-I148,151 and II.147,152 Moreover, a comparison among different metal sites, allowed to verify that the larger electrostatic field of the Mg2+ is able to activate favorably the amine toward CO2 more than any other metal.152 Some similarity with the Rubisco active site where claimed in this case.152
4.4. Atmospheric chemistry
The region of the terrestrial atmosphere extending from altitude between 75 and 110 km is referred as mesosphere/lower thermosphere and is of particular interest because it forms the boundary between the atmosphere and the space. The transition to space is in fact fixed at 105 km (turbopause) because of the change from a bulk to free molecular motion of the gas molecules.165 Mesosphere is also the most sensitive region of the atmosphere to climate change.166 It undergoes to a fast cooling due to the increased presence of molecular radiators like CO2 and methane,167 that have in that region an opposite effect with respect to what is observed in the troposphere (0–17 km). The upper atmosphere is characterized by temperatures between 170 and 250 K and by low pressures (10−6 to 10−3 bar in the mesosphere).165 Because of these peculiar conditions, species (like atomic metallic species) and reactions very close to the gas phase conditions in the molecular quantum mechanics approximation are observed. For example, because of the low pressures, atomic oxygen is the major reactive species present there.165 Also neutral metal atoms and metal ions are present in the upper atmosphere deriving from meteoric ablation. In fact, meteoroids, because of their high entry velocities (tens of km s−1) undergo to rapid frictional heating by collision with air molecules and their constituent minerals subsequently vaporize with formation of atomic metallic species. This process has been estimated to provide daily a mass flux up to 130 t.165,168 Magnesium is the most abundant metallic constituent of meteorites167,169 and, therefore, meteoric ablation injects into the atmosphere large quantities of Mg and Mg+. Systematic observation of global atomic magnesium layers in the atmosphere (Mg and Mg+ species) has become possible only since 2008 through satellite-born UV-visible spectroscopy.165 Before this date, reaction of magnesium with the different reactive species present in the mesosphere were largely investigated in order to individuate the magnesium sink.In the region between 75 and 110 km, magnesium is present in three main forms (see Fig. 10b): Mg+, Mg and Mg(OH)2, concentrated in three different global layers.165 Mg+ layer peaks at heights between 90 and 100 km with a peak ion concentration of (1–5) × 103 cm−3 during daytime, decreasing to 10−2 cm−3 at night.170 The Mg layer exhibits a peak at altitudes around 88 km and lower concentration than the ionic layer,171 for a total Mg+/Mg ratio between 4 and 12, unusually high with respect to what observed for other metals (e.g. Na). These species are fast converted to other molecular species by reaction with highly reactive species as atomic O and H, O3 but also O2, H2O and CO2. The third major gas phase magnesium species, Mg(OH)2, would be formed through the subsequent reaction of Mg with O3 (MgO), O2 (OMgO2) and H2O (see Fig. 10a). Molecular Mg(OH)2 is concentrated in a global layer with a peak at about 85 km and its stability is due to the low reactivity toward atomic O (at difference of oxides and carbonates) and to the low concentration of atomic H at 85 km: in fact, although Mg(OH)2 is destroyed significantly by atomic H, the low ratio [H]/[O] = 1% at 85 km makes the decomposition reaction quite improbable. All the other gas phase magnesium species (MgO, MgO2, OMgO2, MgCO3, …) are short lived and are dynamically and reversibly converted each other (see Fig. 10a and c).165
 | ||
| Fig. 10 (a) Schematic diagram of magnesium chemistry in the upper mesosphere/lower thermosphere region. Major magnesium species are shown in boxes with bold outlines. Important reaction pathways are indicated with thicker arrows.165 Reaction involving CO2 or the products of CO2 reaction with magnesium species are highlighted with blue color. (b) Vertical profiles of Mg+, Mg, and Mg(OH)2 predicted by the 1D atmospheric model for July, at midlatitudes (40°N). These profiles are at midday.165 (c) Optimized geometries (at the B3LYP/6-311+g(2d,p) level of theory) of magnesium-containing molecules likely to form in the upper atmosphere: (i) MgCO3(1A1); (ii) OMgCO3 (3A′′); (iii) Mg(OH)CO2 (2A′); (iv) HOMg(OH)CO2 (1A′). Color legend: Mg (yellow); O (red); C (gray); H (white). Parts (a–c) have been adapted from ref. 165. Reproduced with permission. Copyright 2012 American Chemical Society. | ||
Among the possible reactions of magnesium species in the upper atmosphere, the ones involving CO2 are very short living and are essentially based on neutral magnesium-derived molecular MgO:165
| Mg + O3 MgO + O2 |
| MgO + CO2 + (N2) MgCO3 |
MgOH + CO2, ΔH0 K = −277 kJ mol−1) but the low concentration of atomic H at 80 km would make this reaction less likely to occur.165 Decomposition in atomic O is then the only reaction likely to happen:165| MgCO3 + O OMgCO3 OMgO–CO2 OMgO + CO2 |
| OMgO + CO2 OMgCO3 |
For what concerns Mg+ and hydroxides, reactions with CO2 are not favored with respect to others.165 In particular, Mg+ mostly reacts with O3 giving MgO+ for altitude larger than 90 km.165 For altitude lower than 90 km, Mg+·N2 or Mg+·CO2 complexes will be present, but fast converted to Mg+·O2 (see Fig. 10).165 Reaction of CO2 with hydroxides, on the contrary, is not likely to happens because of the low energetics and the high energetic barrier to the complex rearrangement:165
| Mg(OH)2 + CO2 (+M) HOMg(OH)CO2 |
| MgOH + CO2 (+M) Mg(OH)CO2 |
4.5. Metal surfaces
Sorption of CO2 on pure metallic magnesium is not largely studied being essentially of no practical use. Metallic magnesium is used only as dopant on metallic and oxidic catalyst to enhance their affinity toward CO2. The works present in the literature12,173,174 deal exclusively with single crystal surfaces with the aim to understand as Mg acts as promoter in CO2 activation. The first report of a combined investigation of reactive CO2 chemisorption was at Mg(0001) surface by means of X-ray photoelectron (XPS) and high resolution electron energy loss (HREELS) spectroscopies.12,173 This work showed that CO2 is molecularly adsorbed on Mg only at 85 K; upon increasing the temperature, reactive chemisorptions occurs leading to surface carbonate and oxide. Carbonate was shown to be actually a surface species, whereas oxide is present as underlayer (thickness 0.4 nm). Also for chemisorption on Mg(0001) surfaces, the reaction showed all the characteristics associated with the participation of a precursor state through the Freund–Messmer mechanism (CO2δ−⋯CO2 dimer).124.6. MgO-based oxides
Metal oxides are significantly employed in all the steps of the CO2 cycles, such as for separation, sorption, sequestration and recycling. In particular, their use as catalysts and catalyst supports have been reported, although between the two options, in almost the totality of the cases, they are used as supports of catalytic particles, being both metal and oxidic species. This is due to their large surface area associated to a strong ionicity, that avoid the particles aggregation and then the lowering of the catalytic efficiency. They have also a direct influence on the particles, determining their dimensions and distribution. On the other hand, oxides can show amphoteric, basic, neutral or acidic properties. According to Lewis definition, acidity and basicity indicate electron accepting and electron donating properties, respectively. The choice of the support can then change drastically the ability of the catalyst to activate CO2. Nevertheless, the problem of standardization of the acidity of solids is a difficult one and it has been addressed in the past,61 for example by means of microcalorimetry of adsorption61 and infrared spectroscopy.22 In particular, the possibility to use the average heat of adsorption of NH3 and of CO2 to compare the strength of acid and base surfaces respectively, has been reported.61 In ref. 61, the oxides were all treated at the same temperature (400 °C) because of the importance of surface pretreatment in affecting the relative amount of surface OH (Brønsted acidity), overexposed metal cations (Lewis acidity) and oxygen anions (Lewis basicity) and then the mode of bonding of adsorbed molecules.61 From this statement, the complexity of the problem is clear only partially. In fact, only upon changing the temperature used to activate the material, a direct change in its basicity can be observed.176 Nevertheless, from the comparison of 20 oxides, it was experimentally verified that the acidity is proportional to the charge/radius ratio of the metal (if the oxides are activated at the same temperature). In Fig. 11b, the metal charge/radius ratio is reported as a function of the average isosteric heat of adsorption measured for these 20 oxides. It was evidenced as CO2 affinity towards materials can be used as a measure of their basicity. It is evident that an oxide with a low value of charge/ratio is more ionic in nature and will present more basic sites.176 A higher charge/ratio would correspond to a greater degree of covalency of the oxide and then to a higher acidic character.176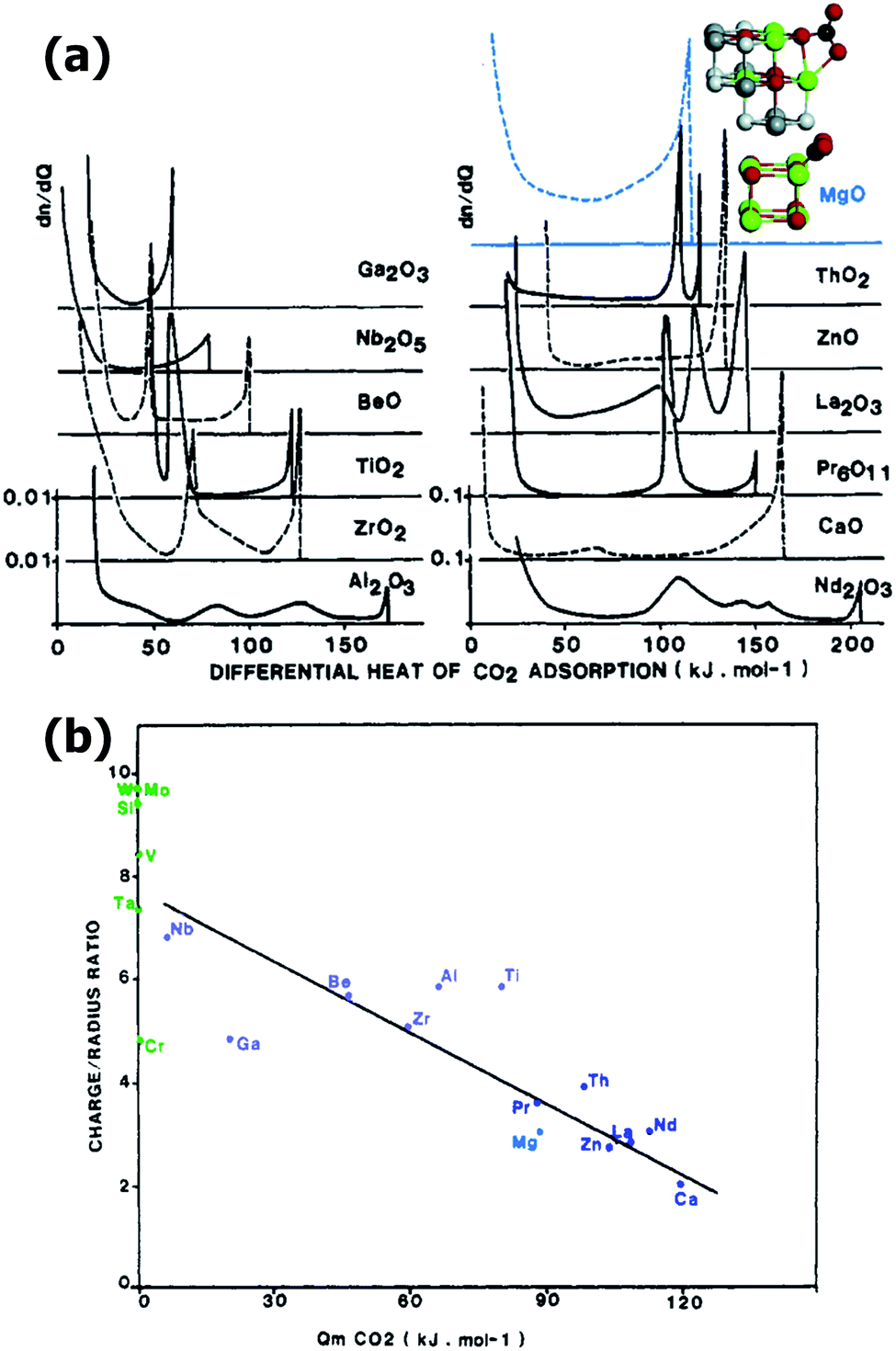 | ||
| Fig. 11 (a) Sites distribution as function of the differential heat of CO2 adsorption as measured at 25 °C on several metal oxides activated at 400 °C as in ref. 61. Cluster representations of monodentate CO2 adsorption on MgO edges and bidentate adsorption on MgO corners (the major species) as obtained at the B3-LYP/6-31G(d) level are also reported. The atoms are represented according to following color code: oxygen (red/white), carbon (black), magnesium (green/grey). (b) Metal charge/radius ratio as function of the average isosteric heat of adsorption for CO2 as in ref. 61. Basic, acidic and amphoteric oxides have been signalized by blue, green and violet color, respectively. (a) and (b) adapted from ref. 61. Reproduced with permission. Copyright 1990 American Chemical Society. Atomistic representations in part (a), adapted from ref. 175. Reproduced with permission. Copyright 2005 American Chemical Society. | ||
Differing on their basic/acidic character, oxides stabilize distinctively the particles and play an active role in the process, determining also the catalytic activity and selectivity.71 Actually, in many processes, the catalytic active site is not represented by the supported particles alone or by the support, but it is at the interfaces between the two species (see Section 2.2).14,178
The broad use of metal oxides in the whole CO2 cycle is due to their ability to form very different bulk and superficial carbonate and bicarbonate species, the relative percentage of which depends strongly on hydration.57 This is exemplified for bulk phase in the ternary phase diagram for the MgO–CO2–H2O system in Fig. 12b. For what concerns superficial species, a picture of the different mechanism of adsorption of CO2 on oxide is given in Fig. 12a. The ability to form these superficial species is particularly important in the activation of CO2 (see Section 2.1) and in determining electron donating properties.12
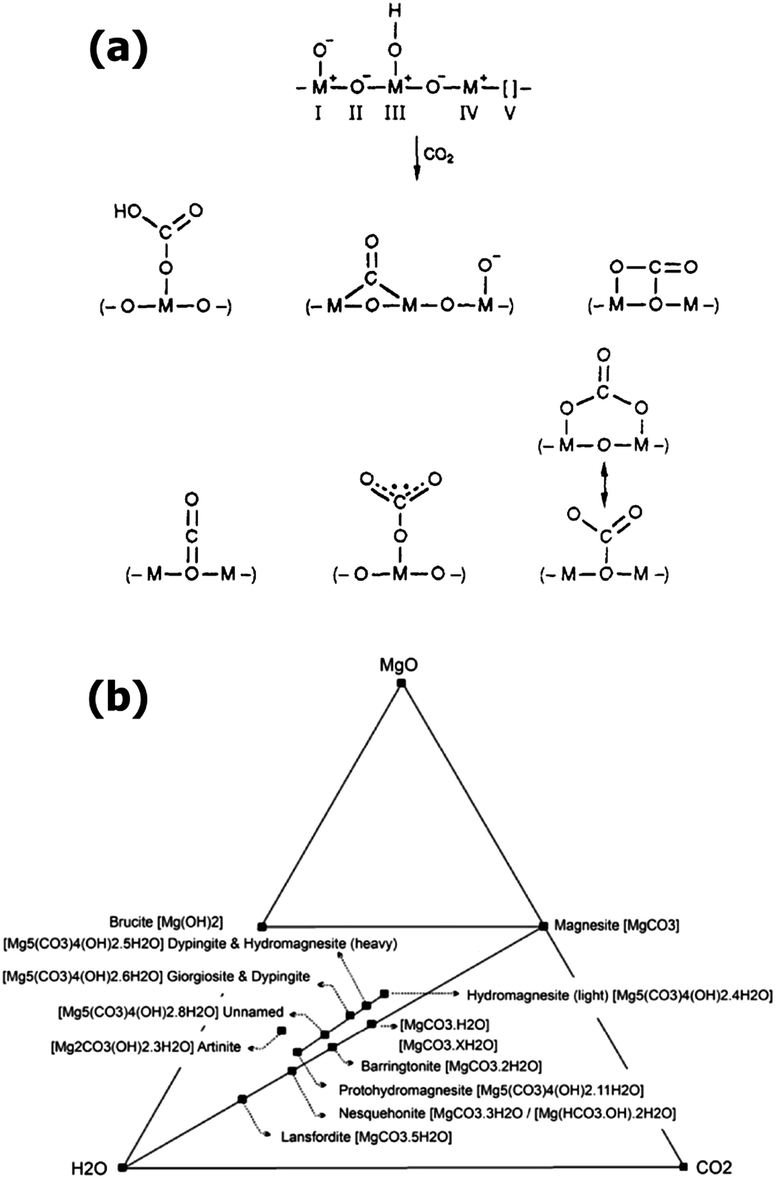 | ||
| Fig. 12 (a) Possible mechanisms of CO2 adsorption on the different types of sites present on the surface of metal oxides. Adapted from ref. 61. Reproduced with permission. Copyright 1990. American Chemical Society. (b) Ternary phase diagram for the MgO–CO2–H2O system with indicated the position of the hydrated and basic hydrates magnesium carbonate. Reproduced with permission from ref. 177. Copyright 2012 Elsevier. | ||
Among basic oxides, MgO is characterized by a medium–high basicity: its CO2 isosteric heat of adsorption is in fact in the middle between CaO (high) and Ga2O3 (low basicity) in Fig. 11a.61 MgO is then expected to facilitate the formation of CO2− species although not stabilizing them too much. CO2 desorption from MgO is observed in fact in two temperature ranges: 60–320 °C, where the most part of the carbonates are decomposed, and 500–800 °C. MgO is an oxide with a rock-salt structure where each Mg2+ ion is octahedrally coordinated to six O2+ and vice versa. For the interaction with a perfect surface constituted by pentacoordinated ions O5c2− and Mg5c2+, it was theoretically shown as175,179 CO2 would be only weakly bound on MgO surface whereas chemically bound with formation of carbonate was observed on a similar surface of CaO. This difference in reactivity is explained by the larger stabilization in MgO of the O2− ions at the surface by the Madelung potential, leading to a lower basicity and reactivity.12
Nevertheless, on the surface of real MgO particles, the landscape is in general more complicated (see Fig. 13a–c). Different irregularities are present that can confer to MgO materials different basicity and reactivity. In particular, the distribution of the lower coordination OLC2− oxides (LC = 3c, 4c for tricoordinated and tetracoordinated ions) and the hydroxyls coverage are important parameters in determining MgO basicity. Activation temperature and synthetic procedures have a drastic influence on these two parameters.180–182 Surface area of MgO strongly varies upon synthesis conditions from 10 m2 g−1 of MgO “smoke” to several hundred m2 g−1 (see Table 6).182 Each OLC2− is accompanied by MgLC′2+ ions, creating a vicinal acid–base couple that can more or less effectively coordinate CO2. The amount of OLC2− sites depends not only on the particle size but also on outgassing the samples at increasing temperature.183 Moreover (see Fig. 13b and c), different OH species are present on the MgO surface, distinguishable between “isolated” OH groups (identifiable by IR spectroscopy as a narrow band at 3749 cm−1 in Fig. 13d) and “linked” OH groups, that is involved in hydrogen bonding (broad band at 3650 cm−1). The hydroxyl coverage can be also changed by increasing the activation temperature or through water exposure.
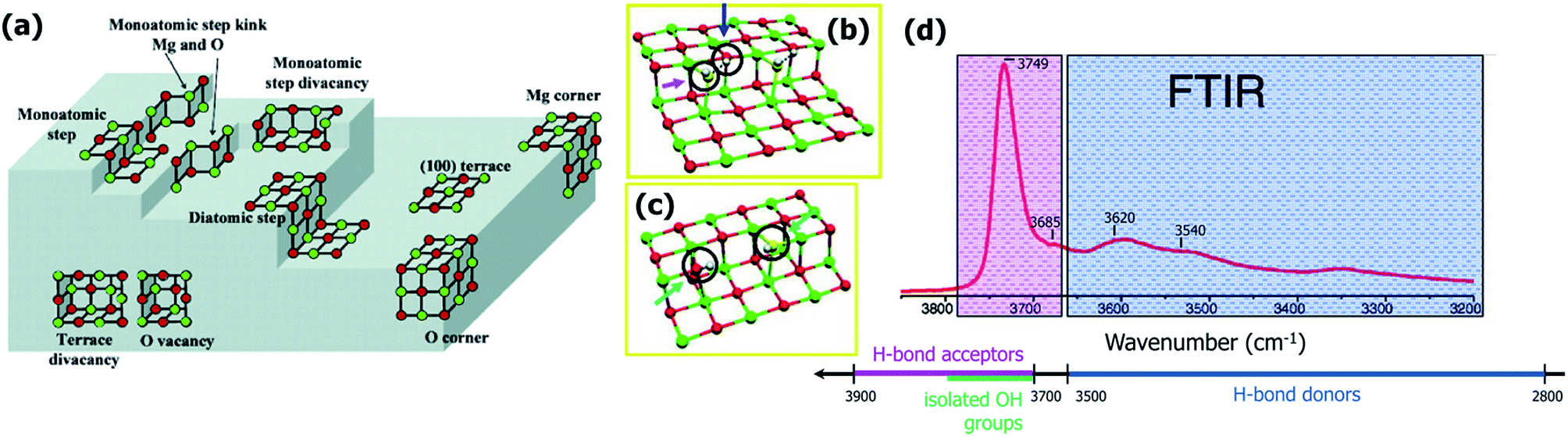 | ||
| Fig. 13 (a) Schematic representation of the irregularities on the MgO surface by using a modified steps and corners model. (b and c) Hydroxyl species on MgO defects. The arrows indicate the different hydroxyl species giving rise to the spectral features in part (d). (d) MgO infrared spectra after activation in vacuum at 300 °C as in ref. 184. Part (a), Adapted with permission from ref. 183. Copyright 2006 American Chemical Society. Parts (b–d) adapted with permission from ref. 185. Copyright 2007 American Chemical Society. The atoms are represented according to the following color code: hydrogen (white), surface oxygen (red), water oxygen (yellow), magnesium (green). | ||
| Material | Precursors | SBETa (m2 g−1) | Vpora (cm3 g−1) | Crystal sizea (nm) | nCO2@P, 1 barb (mmol g−1) | Regeneration T (K) | Cycles | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Surface area and pore volume as obtained from N2 adsorption.b The technique used for the measurement is indicated in parenthesis: vol = volumetry (static conditions), ms = mass spectroscopy, tga = thermal gravimetrical analysis. No physisorbed = CO2 physisorbed in the material is not considered in the reported value because of the operational conditions adopted. | ||||||||
| MgO | Commercial | 0.22–0.45@298 K (tga) | 197 | |||||
| MgO | Nitrate | 7.6 | 0.02 | 40.4 | 0.08@100 °C (vol) | 600 °C | 198 | |
| MgO | Nitrate, CMK-3 carbon | 250 | 0.52 | 1.82@298 K (tga) | 800 °C in N2 (RT-200 °C, >70%) | 3 | 190, 191 and 197 | |
| 2.27@373 K (tga) | ||||||||
| 1.64@298 K (tga, 15% CO2/N2) | ||||||||
| MgO (FM1) | Nitrate, polyethylene oxide | 101 | 0.24 | 10.2 | 1.93@100 °C (vol) | 600 °C in N2 (RT-300 °C, >60%) | 6 | 198 |
| MgO (FM2) | Nitrate, polyethylene oxide | 130 | 0.36 | 9.9 | 2.77@25 °C (vol) | 600 °C in N2 (RT-300 °C, >60%) | 6 | 198 |
| 2.61@100 °C (vol) | ||||||||
| 2.07@150 °C (vol) | ||||||||
| MgO (FM3) | Nitrate, polyethylene oxide | 121 | 0.29 | 10.4 | 2.18@100 °C (vol) | 600 °C in N2 (RT-300 °C, >60%) | 6 | 198 |
| MgO (3DOM MgO) | Nitrate, PMMA, pluronic F127, 20% ethanol | 183 | 0.43 | 5–8 | 0.416@25 °C (vol) no physisorbed | 800 °C in He (RT-400 °C, >50%) | 199 | |
| MgO (3DOM MgO) | Nitrate, PMMA, pluronic F127, 40% ethanol | 243 | 0.45 | 5–8 | 0.568@25 °C (vol) no physisorbed | 800 °C in He (RT-400 °C, >40%) | 199 | |
| (Ca,MgO) (Ca/Mg = 1/50) | 200 (fresh), 38 (after some days in air) | 0.21@25 °C (vol) | 62 | |||||
| MgO–K2CO3 | 1.98@748 K | 748 K | 17 | 56 | ||||
| MgO/C (Mg 21.1 wt%, MgO 42.6 wt%, mPC-MgO-773) | MgCl2, sawdust | 279 | 0.14 | 17.1 | 4.6@353 K (tga) | 200 | ||
| MgO/C (Mg 20.5 wt%, MgO 42.2 wt%, mPC-MgO-873) | Cloride, sawdust | 306 | 0.16 | 18.6 | 2.7@323 K (tga) | 773 in N2 flow | 19 | 200 |
| 5.2@353 K (tga) | ||||||||
| 1.3@573 K (tga) | ||||||||
| MgO/C (Mg 19.4 wt%, MgO 41.4 wt%, mPC-MgO-973) | Cloride, sawdust | 298 | 0.15 | 17.8 | 5.1@353 K (tga) | 200 | ||
| MgO/γ-Al2O3 (24 wt% MgO, 5 A3M) | Nitrate, Al(NO3)3, P123 polymer | 202 | 0.48 | 1.02@200 °C (10% CO2, 90% N2, tga-ms) | 600 °C in Ar flow (RT-350 °C, >50% for dry CO2 and >70% for wet CO2) | 6 | 192 | |
| 2.11@200 °C (10% CO2, 80% N2, 10% H2O, tga-ms) | ||||||||
| MgO/γ-Al2O3 (39 wt% MgO, 5 A5M) | Nitrate, Al(NO3)3, P123 polymer | 177 | 0.45 | 1.75@200 °C (10% CO2, 90% N2, tga-ms) | 600 °C in Ar flow (RT-350 °C, >50% for dry CO2 and >70% for wet CO2) | 6 | 192 | |
| 2.98@200 °C (10% CO2, 80% N2, 10% H2O, tga-ms) | ||||||||
| MgO/γ-Al2O3 (55 wt% MgO, 5 A7M) | Nitrate, Al(NO3)3, P123 polymer | 108 | 0.26 | 0.84@200 °C (10% CO2, 90% N2, tga-ms) | 600 °C in Ar flow (RT-350 °C, >50% for dry CO2 and >70% for wet CO2) | 6 | 192 | |
| 1.91@200 °C (10% CO2, 80% N2, 10% H2O, tga-ms) | ||||||||
Contrary to what expected on the basis of its complicated surface structure,175,181,183,185 microcalorimetric measurements of carbon dioxide conducted at 25 °C on a high surface area MgO (197 m2 g−1, activation temperature 400 °C)61 indicate an homogeneous adsorption behavior with two plateau at low (0–0.6 μmol m−2) and high (>1.6 μmol m−2) coverage at 110 and 15 kJ mol−1 respectively, with a monotonic decrease for intermediate loadings. Accordingly, it has been shown by infrared spectroscopy studies186 as chemisorption of CO2 on MgO reveals mainly bidentate adsorbed species with a little part of unidentate. Quantum mechanical calculations suggest that monodentate species would be formed on edge sites (4c) and bidentate on corner sites (3c) of MgO (see Fig. 11a).175
Several mesoporous MgO materials have been reported in literature, synthesized for example from sol–gel syntheses,62 through the use of surfactants or by structure replication of mesoporous carbons190, 191 and polymers.192 Nanosized crystal MgO have been also reported, synthesized through the use of surfactants,62,193 hypercritical sol–gel drying194 or MOF decomposition in air at high temperatures.195,196 In these cases, higher CO2 capacities were obtained with respect to commercial MgO (0.2–0.5 mmol g−1,62 see Table 6) although still far from the stoichiometric capacity (24 mmol g−1). This increase is related to the higher surface/volume ratio of the particles and then to the higher number of tetracoordinated and tricoordinated sites on the MgO surfaces, besides the fact that the smaller dimensions make the uptake less dependent on the diffusion of CO2 in the bulk of the particles. MgO doping with other oxides does not bring significant increase in the CO2 uptake (about 0.5 mmol g−1).62,63
Unfortunately, mesoporous and nanosized MgO fastly lose their capacity upon cycling. The sintering has been proven to rely on the necessity to heat up the system relatively fast.194 Very slow heating rate would be necessary during the regeneration, that are impracticable in a plant. In these respect, a possible solution has been reported to be to support MgO on high surface area materials as zeolite,201–203 mesoporous carbons,200 alumina192 and silica materials (SBA-15).204 One of the most interesting results in this area has been reported by Liu et al.200 MgO nanoparticles were in fact synthetized through fast pyrolysis of biomass waste (sawdust) preloaded with MgCl2. A maximum capacity of 5.45 mmol g−1 at 80 °C and 1 bar was obtained with a good cyclability and a low regeneration temperature.200 This synthesis is particularly appealing being MgCl2 largely abundant in seawater.
Magnesium oxide has been also used as support for other CO2 sorbent type as dry carbonates (see Section 2.2.4) with very low performances67 and for CaO. Calcium looping technologies are at present one of the most efficient technologies for CO2 capture between high temperature CO2 sorbents,63,64 especially if applied to cement industry.65 Unfortunately, CaO suffers severely from sintering during regeneration.63 MgO has been successfully used as supporting material for enhancing the sintering resistant properties of CaO.63,205 If a CaO/MgO material with CaO/MgO = 0.4 is adopted, the starting CO2 capacity of 15 mmol g−1 was retained also after 50 cycles in a 100% CO2 flow at 750 °C.205
Hydrogenation reactions. MgO,14,207 Mg/SiO2,14,32,208 and MgAl2O4 (ref. 14 and 207) have been tested as support for Ru and Pd particles in catalysts both for CO2 methanation (reaction [2] in Table 1) and reverse water gas shift (reaction [1]) reactions, for their ability to form carbonates and to strongly interact with the metal particles (see Section 2.2.1). The turnover frequency for CO2 methanation obtained for high metal dispersion on these systems is in the order: Ru/Al2O3 (16
500 s−1) > Ru/MgAl2O4 (8800 s−1) > Ru/MgO (7900 s−1) > Ru/C (2500 s−1),207 indicating as the stronger the interaction with CO2, the faster the reaction kinetics. The apparent activation energy for MgO-based materials for CO2 methanation has been also reported for Ru/MgO (74 kJ mol−1)209 and Pd/Mg/SiO2 (82.2 ± 0.2 kJ mol−1).32 This value was found to strongly decrease upon mechanical milling of the catalysts:209 after vibrating milling the activation energy on Ru/MgO decreases to 41 kJ mol−1 and an even lower value was obtained for Fe–Ni/MgO (39 kJ mol−1).209It was reported in Section 2.2.1 as small changes in the catalyst composition can also favor one of the hydrogenation reactions over the others. For example, it was found that adding a small amount of Mg to the silica precursor (amorphous Mg/SiO2 oxide) would favor the formation of methane over CO in Pd based catalyst by stabilizing carbonate species on the catalyst surface and then methanation over RWGS reaction.32 The ability to create stable carbonates is at the basis of the larger yields obtained by Al2O3 with respect to most part of oxides. On the contrary, MgO based catalysts are not largely used being easily poisoned by small pollutants in the gas feed (e.g. H2S) more easily than other supports.14
CO2 methanation mechanism has been computationally and experimentally investigated by Kim et al.208 on a Pd–MgO/SiO2. This study confirmed the dual site reaction mechanism for methanation/RWSG reactions. It emerged as MgO initiates the reaction by binding CO2 molecules with formation of an activated surface magnesium carbonate species. Pd species would dissociate hydrogen molecules that then hydrogenate the carbonates and residual carbon atoms. This mechanism agrees on qualitative expectations and on what often proposed on the basis of experimental evidences.32
Dry reforming of methane. Basic oxides, and in particular MgO, are used as supports or as additives in metal-based catalysts for methane reforming because they enhance the mechanical strength of the catalyst42 but, even more important, they lower the coking rate,28 that represent the main problem in this sector (see Section 2.2.2). Contemporaneously, they promote the activation of carbon dioxide, which both participates to the reaction and to the oxidation of coke.28 MgO is an irreducible oxide and this guarantees a larger stability of the catalysts if used as support. For all these reasons, Ni-based catalysts obtained by coprecipitation of aluminium oxide and magnesium oxide, although proposed for the first time by Rostrup-Nielsen in 1974,210 are still under investigation.211 Nevertheless, the propensity of MgO–NiO to form solid solution is well known. This fact markedly affect the catalityc activity of Ni/MgO catalysts. In fact, if the calcination temperature and the Ni dispersion facilitate the diffusion of Ni2+ ions in the MgO lattice, solid solutions are formed with the result that the Ni species would not be reducible anymore.42 Too small Ni particles (<100 Å) have been also to be avoided in these catalysts because too strong Ni–O bonds are formed with the support, resulting in lower activities and more sensitivity to coke deposit. The Boudouard reaction would be also inhibited only on larger Ni particles.
MgO has been also used as support for Rh and Ru-based catalysts with more promising results with respect to other supports.28 The dependence upon CO2 on the kinetics of the reaction increases with the basicity of the support being important for Rh/MgO system instead of what observed for Al2O3 supported catalyst where no dependence upon CO2 concentration was observed.45 In fact, Rh/Al2O3 resulted to have a higher reactant conversion with respect to (in the order) Rh/TiO2, Rh/SiO2 and Rh/MgO.45 Nevertheless, Rh/MgO showed the highest stability (100 h)212 because of the more effective carbon oxidation by CO2 due to the higher basicity of the support. Completely different was the behavior for Ru-based catalysts, where at 1 bar, 2%Ru/MgO resulted the catalysts with the highest activity with respect to corresponding TiO2, SiO2 and Al2O3 supported system and a lower coke formation.213
Mg has been also used as promoter in dry methane reforming catalysts. The effect of the promoter was found to be strongly dependent upon the ratio of the additive to the active metal.28 For example, studies of Na, K, Mg as additive in Pt/Al2O3,214 indicated as Mg causes the highest increase in the activity with respect to the doped Na and K catalysts for 0.02% and 0.1%Pt/Al2O3 systems, whereas for 0.5%Pt/Al2O3 the K-doped systems showed higher performances. Nevertheless, for the 0.5%Pt/Al2O3 no deactivation was observed in any case, suggesting also a difference in the particle aggregation.214
Synthesis of methanol. Two are the main common problems for catalysts to be used for the synthesis of methanol: Cu clustering and carbon deactivation (see Section 2.2.3). Perovskites are oxidic materials with a ABO3 formula (A is the cation having the larger dimensions and B is the smaller one, e.g. CaTiO3). They are characterized by interesting properties as high thermal stabilities and large oxygen mobility. The latter is a characteristic particularly interesting in the applications concerning CO2 reactions, because it would facilitate coke oxidation and then increase the stability of the catalysts. The perovskite structure can be seen as an AO structure hosting the B ion. In the use of perovskites as catalysts, B constitutes the catalytic active site that results in this way very well dispersed.40 For these reasons, although the surface areas of these materials are very low (0.5–2 m2 g−1), they allow to obtain large metal dispersion accompanied by a large thermal stability toward sintering. Perovskite type A2BO4 consist of alternating layers of ABO3 perovskite and AO rock salt which exhibit variable oxygen stochiometry.40 For their particular oxygen structure, A2BO4 allowed the stabilization of Cu species with special valence.40 La2CuO4 perovskite catalysts for methanol synthesis40 have been prepared by sol–gel synthesis. It was observed as the addition of Ce, Mg and Zr leads to several benefits as a remarkable decrease in particle dimension, lowering of the reduction temperature, an increase in the Cu dispersion and in the basic sites. These catalysts have been reported to show high selectivity for methanol.40 Also high turnover frequency and long term stability in operation conditions (312 h) were obtained.40 For what concerns the Mg-doped material (La0.8Mg0.2Cu0.7Zn0.3Ox), high dispersion of Mg was reached (MgO particles were no detectable by XRD) because it can easily substitute Cu2+ and Zn2+ in the B site. On the reactivity point of view, the Mg based material showed the highest selectivity toward CH3OH formation (65.2%) with respect to the pristine material and to the modified Ce and Zr ones.40
4.7. Hydroxides
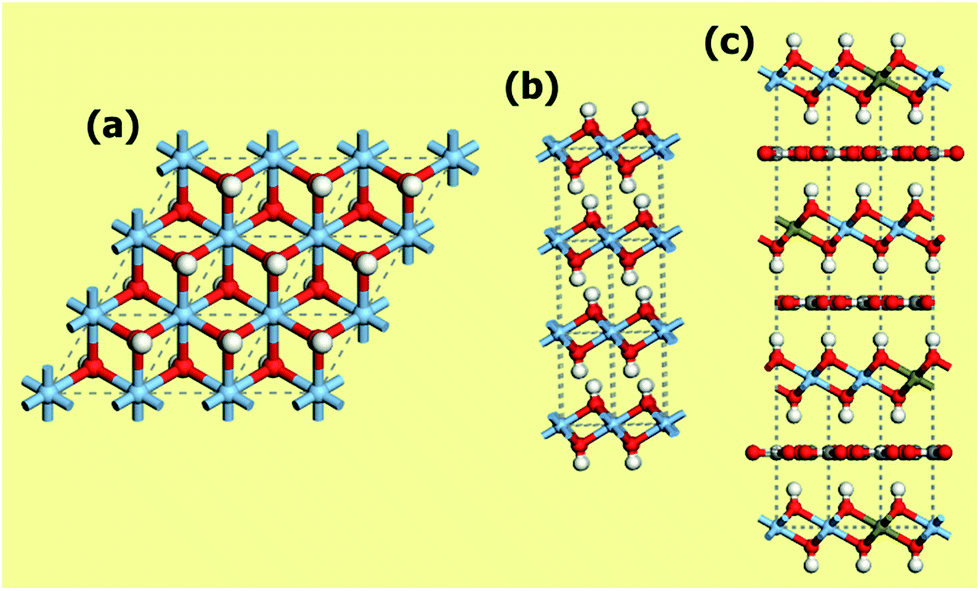 | ||
| Fig. 14 (a) Top view of the brucite layer in Mg(OH)2 and (b) side view of Mg(OH)2 layers as in the structure reported in ref. 215. (c) Side view of hydrotalcite layers in the structure reported in ref. 216. The atoms are represented according to the following color code: hydrogen (white), oxygen (red), carbon (grey), magnesium (light blue), aluminium (dark green). | ||
Mg(OH)2 is the product of hydratation of MgO and it is characterized by a lower basicity than MgO.176 For the disposal and separation of large amounts of CO2 through the formation of carbonate, the carbonation of Mg(OH)2 is of particular interest because of the relative easiness with it can be precipitated from sea water and dissolved minerals, and because it occurs naturally as brucite.58 The carbonation process in Mg(OH)2 is governed by the reaction:58
| Mg(OH)2 (s) + CO2 (g) MgCO3 (s) + H2O (g) |
| Mg(OH)2 (s) MgO (s) + H2O (g) |
The kinetics of simultaneous dehydroxilation and carbonation of precipitated Mg(OH)2 were studied by Butt et al. at 0.78 bar.58 This Mg(OH)2 sample was constituted by dense agglomerates of relatively spherical particle 28.7 μm in diameter formed by submicrometer-sized single crystals.58 Dehydroxylation was conducted both in helium and in helium/CO2 flow (He:CO2 = 1:2). An activation energy of 146 kJ mol−1 was determined for dehydroxylation in pure helium in the 375 < T < 475 °C range.58 The same reaction in vacuum was reported to have an Ea of only 53–126 kJ mol−1.58 The mechanism of dehydroxylation was also determined: it appeared as a nucleation and growth process, accompanied by extensive crystallite cracking with the formation of a porous, pseudomorphic structure comprised of MgO crystallites of the order of 100–200 Å. Dehydroxylation in He/CO2 flow, corresponds to the reaction [9] in Table 1, that is to Mg(OH)2 carbonation. This reaction in this system has an Ea about three times the one for the reaction in absence of CO2 (304 kJ mol−1). Carbonation was determined to start at 275 °C, reaching the most rapid carbonation kinetics near 375 °C and ending at 450 °C. During carbonation, MgCO3 precipitates on the surface of the crystals as 200 × 750 Å particulates acting as a passivating layer hindering both the outward diffusion of H2O and the inward diffusion of CO2. This layer would be then the cause of the increase in Ea observed for the reaction in He/CO2 flow with respect to He flow. This experiment also validated the hypotheses to explain the low yields observed in carbonation of MgO. Correspondingly, the kinetics of reaction are very slow. In fact, after 12 h of reaction, the carbonation fraction was only between 6.7 and 16.7 wt% for isothermal reactions conducted at temperatures between 250 and 500 °C. Interestingly, this needs that a significant amount of the specimen remains carbonated also at temperature higher than the dissociation temperature of MgCO3 (385 °C) in presence of high concentration of CO2 gas in the flow. This observation indicates the suitability of this system for PSA process: at T > 385 °C, it would be sufficient to lower the CO2 concentration in the feed to regenerate the adsorbent. Nevertheless, the presence of the passivating layer limits the CO2 uptake to about 15% of the stoichiometric capacity at 250 °C and 500 °C and to the 37% at 375 °C.
An increase in the temperature window of reactivity down to 200 °C and a slight improvement in the absorption capacities of Mg(OH)2 were obtained by dispersing it in amorphous silica by hydrolyzing a solid mixture of magnesium hydroxide and sodium orthosilicate, followed by simply drying at 100 °C for 1 h.30 XRD pattern conducted on the fresh material indicated the presence of Mg(OH)2 and impurities of NaOH and Na. The siliceous fraction of the sample, being amorphous, was not detectable. Thermodynamic equilibrium data acquired for this system for the decomposition of 1 mol of MgCO3 in the presence of 1 mol of H2O at 1.01 and 20.3 bar are reported in Fig. 15a and b, respectively.30
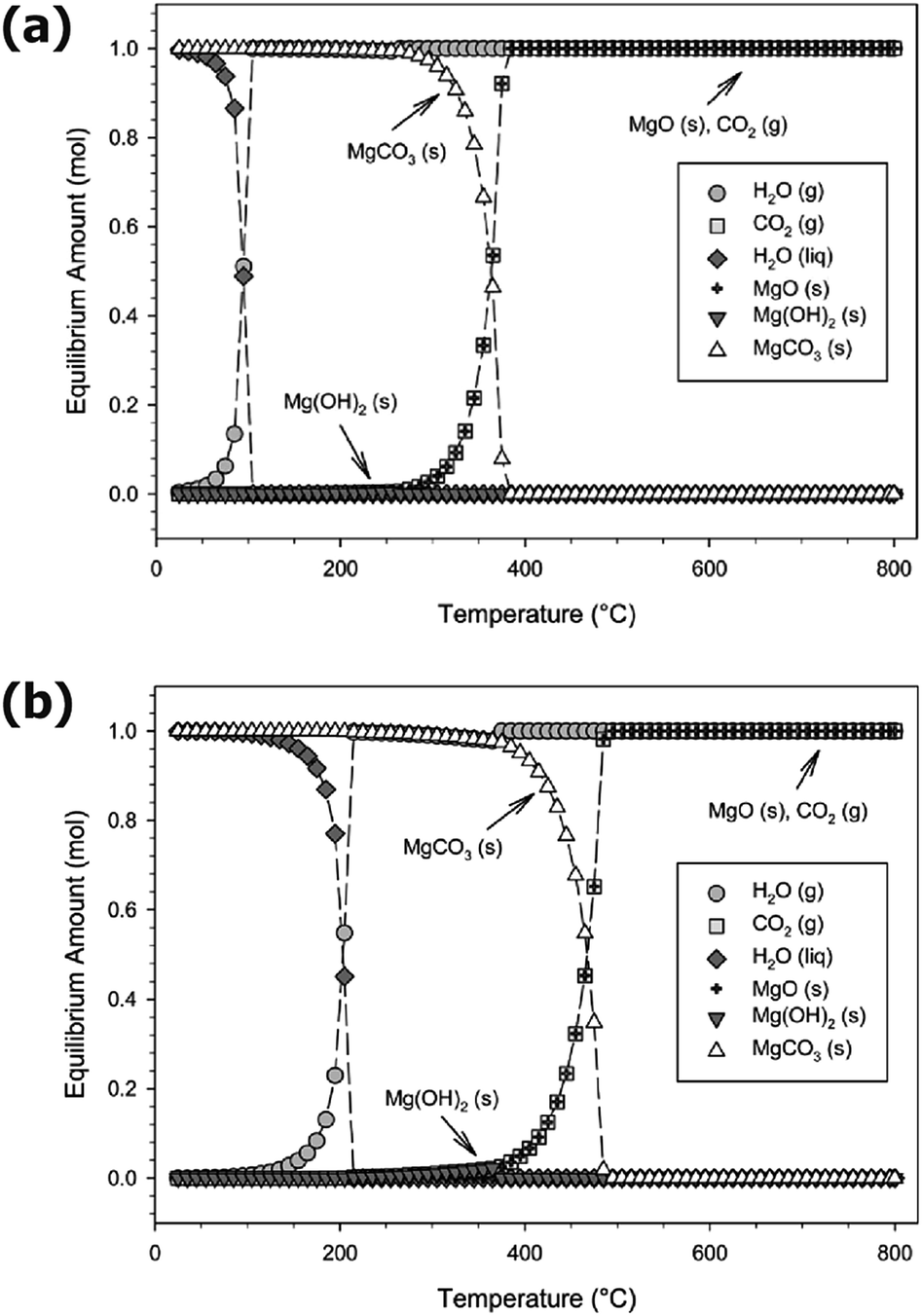 | ||
| Fig. 15 Thermodynamic equilibrium analysis as a function of temperature for a system of 1 mol of MgCO3 (from Mg(OH)2/SiO2 system), 1 mol of H2O, and 0.1 mol of N2 at a pressure of (a) 1.01 and (b) 20.3 bar. Reproduced with permission from ref. 30. Copyright 2009 American Chemical Society. | ||
This Mg(OH)2/SiO2 system is an excellent candidate sorbent for CO2 removal under integrated gasification combined cycle (IGCC) conditions (200–315 °C).30 The compound has in fact a considerably higher CO2 capture capacity than that of the commercial Selexol® process (3–4 versus 0.3 mmol g−1),30 and the energy required for the reverse carbonate decomposition was verified to be significantly lower than for other processes.30 The system was fully regenerated for temperatures starting from 375 °C at 20 bar, also in presence of water. Such a small difference in the temperatures for CO2 removal and absorbent regeneration is particularly interesting for applications. High pressures for regeneration were considered in the study of Siriwardane et al.30 because they allow to save the CO2 compression costs required for CO2 sequestration. The presence of water during regeneration was actually essential in order to allow a higher degree of rehydroxylation, condition necessary to guarantee high CO2 capture and cyclability of the material. In fact, MgO at 200 °C and 20 bar (CO2/Ar/H2O/N2 = 28/28/10/34) showed uptake of only 0.25 mmol g−1 against 2.25 mmol g−1 of Mg(OH)2–SiO2 in same conditions, despite the higher carbonation enthalpy of MgO. These results suggest that the kinetics of CO2 capture over MgO are much slower than those over Mg(OH)2. Therefore the presence of Mg(OH)2 is necessary for the CO2 process in conditions interesting for IGCC. Moreover, sorbent capacity was observed to increase during cycles. This was associated to an increase degree of rehydroxylation. Nevertheless, the importance of Mg(OH)2 to carbonation can appear as a nonsense being the reaction of carbonation preceded by the dehydroxylation of the material. Moreover, the dehydroxylation is a process strongly endothermic and then cannot help locally the carbonation reaction, if not even hinders it. In my opinion, the beneficial effect of the presence in the starting material of Mg(OH)2 is related to the different nanostructure of the MgO that is formed. In particular, repeated cycles of hydroxylation – de-hydroxylation would help keeping MgO as a finely divided, highly reactive, material. In fact, as verified by Butt et al.58 the Mg(OH)2 dehydroxylation is a nucleation and growth process and the formed MgCO3 crystal are significantly smaller than the starting Mg(OH)2 ones and concentrated only on the surface. The carbonation reaction has been verified by Butt et al.58 to happen during the dehydroxylation. Likely, the surface of the formed nuclei of MgO outside the Mg(OH)2 crystals are very similar to the 3c species reported to exist in very special position on surface of bulk MgO (see Section 4.6). The high defectivity of the formed MgO would facilitate the activation of CO2 and then the carbonation reaction of the material surface. This would also be another reason for the superficial nature of the carbonate formed, besides to rely on CO2 diffusion problems. Moreover, it was found as CO2 removal efficiency was higher if the regeneration was conducted at higher pressures (10 instead of 1 bar).30 This was explained by the fact that significant rehydroxylation of MgO to Mg(OH)2 was observed at T = 300 °C at 20 bar and T = 275 °C at 10 bar, that is increasing the pressure increase the stability of the hydroxide and then the temperature window for the rehydroxylation process. Interestingly, CO2 capture in Mg(OH)2–SiO2 was observed up to 300 °C at 1 bar and up to 400 °C at 30 bar in a CO2/Ar/H2O/N2 flow (10–30% H2O, 10–30% CO2).
XRD analysis conducted30 on Mg(OH)2/SiO2 after exposure to CO2 at 200 °C and 20 bar, indicated a sample only partially carbonated being the pattern still dominated by Mg(OH)2 peaks, confirming that the capacity of the system was not fully exploited. Regeneration of the system at 400 °C and 20 bar caused the formation of MgO, constituting the larger fraction of the sample, with some residual MgCO3 and the appearance of forsterite (Mg2SiO4) and Na2SiO3 features.
The fact that the absorption capacity of Mg(OH)2 also in Mg(OH)2–SiO2 system was not fully exploited is likely imputable to its low surface area (2.4–3.0 m2 g−1).30 An increase in the carbonation capacity can be reached by dispersing Mg(OH)2 on high surface area materials, as zeolites (see Table 7).217 Hong et al.217 precipitated Mg(OH)2 from a MgCl2 solution on 13X and CaCHA zeolites and on a mesoporous CaCHA sample (CaCHA(M)).217 X-ray diffraction showed that the pattern of 13X was maintained after the impregnation, whereas for the CaCHA samples only the peaks associated to Mg(OH)2 were observable, likely because a structure collapse due to the use of NH4OH in the hydroxide synthesis. An uniform distribution of Mg2+ was observed for all the samples. For 13X, scanning electron microscopy images showed the formation of Mg(OH)2 nanoplatelets on the external surface of the zeolite.217 For this material, 99% of the hydroxide was carbonated at 200 °C and 10 bar (CO2:H2O:N2 = 30:10:60) whereas only 12.5% of pure Mg(OH)2 was reacted in the same conditions (2.16 mmol g−1).217 At 20 bar, 99.9% of the supported hydroxide was reacted (see Table 7).217 Interestingly, although the zeolite structure of 13X–Mg(OH)2 collapsed after only 4 cycle, the CO2 capture capacity was maintained because mainly related to the dispersion of Mg(OH)2.217
| Material | Si/Al | Framework type | SBETa (m2 g−1) | SLangmuira (m2 g−1) | Vpora (cm3 g−1) | nCO2@P,Tb,c (mmol g−1) | qiso (kJ mol−1) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a Surface area and pore volume as obtained from N2 adsorption.b The technique used for the measurement is indicated in parenthesis: vol = volumetry (static conditions), ms = mass spectroscopy, tga = thermal gravimetrical analysis.c Conversion from original data in wt% made on the assumption that wt% = CO2 weight/(CO2 weight + sample weight) × 100, if not otherwise specified.d Zeolite synthesized in presence of [3-(trimethoxysilyl)propyl]octadecyldimethylammonium chloride (TPOAC; 72%, Aldrich), a mesopore forming agent.e Measured in presence of water in the inlet gas (CO2:N2:H2O = 3:6:1). |
||||||||
| LS Mg-KFI (Mg11Li4.5Na0.7K2.8((CH3)4N)6 [Si60Al36O192]) | 1.67 | KFI | 337e | — | 0.14 (Micropore) | 0.20@303 K and 0.9 mbar (vol) | — | 236 |
| 3.09@303 K and 1 bar (vol) | ||||||||
| 3.64@303 K and 35 bar (vol) | ||||||||
| Mg-ZK-5 | 4.7 | KFI | — | — | 0.21 | 4.43@303 K and 1 bar (vol) | 30.8–34.7 | 233 |
| Mg-CHA (Na3.3Mg3.8 [Al10.9Si25.1O72]) | 2.4 | CHA | — | — | — | 4.28@273 K and 1.03 bar (vol) | 30–41 | 234 |
| MgCHA | 2.4 | CHA | 503 | 0.38 | 4.57@298 K and 1 bar (vol) | 217 | ||
| CaCHA-Mg(OH)2 (15 wt%) | 2.4 | CHA | 610 | 0.65 | 0.57@473 K and 1 bar (tga)e | 217 | ||
| 1.14@473 K and 20 bar (ms)e | ||||||||
| CaCHA(M)-Mg(OH)2 (Mg(OH)2 = 18 wt%)d | 2.4 | CHA | 173 | 0.57 | 0.45@473 K and 1 bar (tga)e | 217 | ||
| 1.75@473 K and 20 bare (ms) | ||||||||
| Mg-A (Na0.48Mg0.26AlSiO4) | 1.0 | LTA | — | — | — | 3.89@298 K and 1.03 bar (vol) | 34–36 | 76 |
| Mg-X (Na0.38Mg0.31AlSi1.18O4.36) | 1.1 | FAU | — | — | — | 5.00@298 K and 1.03 bar (vol) | 33–40 | 76 |
| 13X-Mg(OH)2 (8 wt%) | 1.4 | FAU | 750 | 0.65 | 0.55@473 K and 1 bare (tga) | 217 | ||
| 2.11@473 K and 20 bare (ms) | ||||||||
| 13X | 1.4 | FAU | 804 | 0.39 | 5.9@298 K and 1 bar (vol) | 49 | 76 and 217 | |
| 0.48@473 K and 1 bare (tga) | ||||||||
| 0.70@473 K and 20 bare (ms) | ||||||||
| Mg(OH)2 | — | — | ∼2 | 2.99@648 K and 0.5 bar | 58 | |||
Moreover, LDH properties can be easily tuned by changing, among the others, the composition of their structure and the nature and the number of counterions (by changing the M2+/M3+ ratio). In particular, as other layered materials (e.g. clays, Section 4.8.2), the accessibility of the interlayer space is strongly dependent on the dimensions of the species hosted in the interlayer. The swelling can be facilitated as in clays by the use of counterions of appropriate size.220 Hydrotalcite surface area was varied by Fetter et al.221 in the range 20–350 m2 g−1 by changing the chemical composition. Wang et al.63 recently reviewed the properties of LDH as CO2 sorbents and stated that they represent the most important intermediate temperature (200–400 °C range) CO2 sorbents, that is for precombustion processes.11 Among the possible modifications aimed to tune their CO2 capacities, intercalation of organic anions is particularly promising. For example, the use of stearate in Mg–Al LDH allowed to increase the CO2 uptake from 0.5 (for Ax/mm− = CO32−) to 1.25 mmol g−1 at 200 °C and 1 bar.220 This because the long chain hydrocarbon actually exfoliated completely the system, allowing to maximize the surface exposed to CO2. Also graphite oxide has been used as counterion in Mg–Al LDH with beneficial effects on both sorption capacities and recyclability.222
Since their composition, it is evident that LDH present intrinsically anionic and basic sites and for this reason they are particularly suitable to activate carbon dioxide. Their acidic–basic properties can be modulated by changing the compensating anion, the amount and the nature of structural cations. For example, enhanced CO2 adsorption properties have been reported if Al3+ is partially substituted by Ga3+.223–225
Mg–Al hydrotalcites have been also used as support for Ru–Ni particles for dry methane reforming. Mg–Al hydrotalcites are very good reforming catalysts as they resist to carbon formation because of their basic properties, high surface area and thermal stability.226
Calcination of hydrotalcites allow to obtain mixed Mg–Al oxides having weak acid and basic properties that can be tuned by changing the calcination temperature.52 In fact, the calcination temperature has a strong influence on the number of active Mg–O species created and on the material structure (crystalline or amorphous).63,227 Because of the presence of both basic and acidic properties (see Section 2.1), also these oxides have been tested as heterogeneous catalysts for CO2 reactions and in particular in the direct carboxylation of methanol by Stoian et al.52 In order to increase the number of active sites, the hydrotalcites (Mg/Al = 3) were in turn supported on high surface area materials such as silica lyogels (uncalcined silica xerogels, 1000 m2 g−1) before the calcination at 450 °C (4.4 wt%). Surface areas as high as 646 m2 g−1 were obtained for the supported material. Both supported and unsupported Mg–Al oxide showed very high selectivity for temperatures comprised between 90 and 130 °C (99.9%, CO2/CH3OH = 25). For its performances, this material represents an important breakthrough in this research field. At higher temperatures (>130 °C), an increase in the formation of dimethyl ether as side product was observed mainly related to thermal decomposition of the formed dymethyl carbonate on the same basic/acid sites that have allowed its formation.52 The conversion to dimethyl carbonate at 130 °C amounted to only 1.8 and 15.9% for the unsupported and supported mixed oxide, respectively.52 Both these values are still higher than those reported for all the previously reported heterogeneous catalysts for this reaction (about 1%).228 Stability of the catalyst up to 480 h was also reported.52 On further increasing of the temperature (175 °C), dimethyl ether resulted the almost total product of the reaction (95%). This result is quite interesting too because, as stated in Section 2.2, dimethyl ether can directly substitute diesel oil in internal combustion engines.14
4.8. Silicates
Silicates are the largest group of mineral compounds. They constitutes well over the 90% of rocks forming minerals on the Earth's crust. Several synthetic structures have been also reported, belonging to the silicates group, further enlarging their number.Since their name it can be inferred that at the basis of their structures there is silicon that is present in [SiO4]4− tetrahedra. Because of the flateness of the energy potential surface on the rotation of two [SiO4]4− units, several structures can be obtained by using this tetrahedron as building unit (see for example zeolites, Section 4.8.1). In addition, in silicate structures other elements can be present, further enlarging the number of possible frameworks. Magnesium is often hosted in silicate frameworks, occupying the central position of octahedral building units (see Fig. 17). Magnesium can be also present in silicates as an extraframework species for example in zeolites and in clays, where it is hosted in the material pores, to counterbalance the negative charge of the frameworks. Silicates have been largely investigated for their affinity towards CO2.55,60,229 The nature of the interaction with CO2 in sorption and separation processes for silicate materials strongly depends on the subgroup considered, going from molecular adsorption in zeolites and clays materials (Sections 4.8.1 and 4.8.2) to chemical reaction (Section 4.8.3).
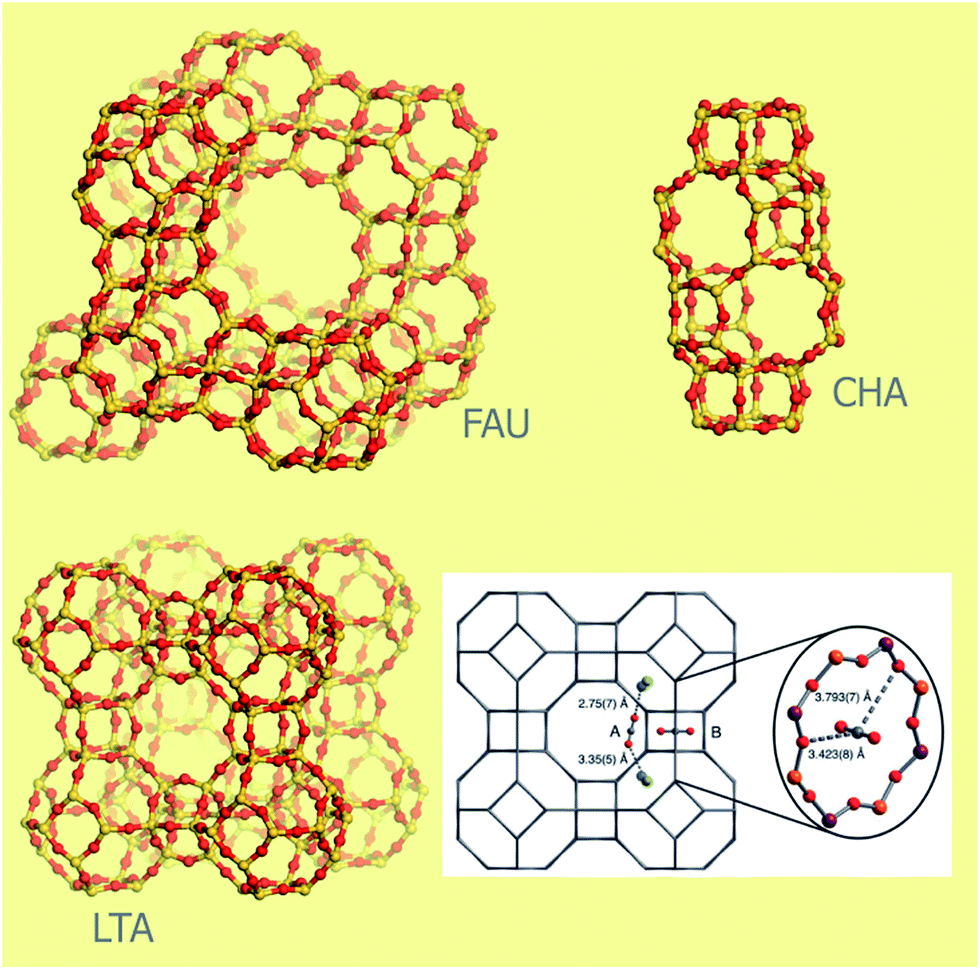 | ||
| Fig. 16 Zeolitic structures. The largest cavities (or supercages) of zeolite A (LTA framework), chabazite (CHA) and zeolite X or Y (FAU) are reported. The atoms are represented in the color code: oxygen (red), silicon/aluminium (yellow). The cations are omitted for clarity. In the scheme, the structures associated with CO2 adsorption at two sites in zeolites Ca-A (A and B) are represented as determined from neutron powder diffraction measurements at 10 K. Gray, red, blue, green, orange, and purple spheres represent C, O, Na, Ca, Al, and Si atoms. Reproduced with permission from ref. 76. Copyright 2013 Royal Society of Chemistry. | ||
Zeolites having a porosity constituted by cavities with access apertures of 3.5–4.5 Å (that is close to the CO2 kinetic diameter) yield the highest recorded selectivities for both CO2/N2 and CO2/CH4 separation.78,233 This pore dimension corresponds for example to siliceous 8-rings, that are typical of the CHA,234 LTA235 and KFI233,236 frameworks. Because of their high working capacities, 13X (NaX) and NaY zeolites are considered the best CO2 adsorbents but only when low pressure feed and low regeneration pressures are used.233 Nevertheless, because of their larger pore windows (12-rings) their CO2/N2 selectivity is not high enough to allow an effective CO2 adsorption from a high purity CO2 stream.233
Some of the magnesium zeolites studied in literature for CO2 adsorption are listed in Table 7. It is evident that in the zeolite formula other cations than Mg are present. In fact, often zeolites are synthesized in their sodium form and the corresponding magnesium zeolite is obtained by cation exchange by stirring the zeolite in an aqueous solution of a magnesium salt. Exchange of monovalent with divalent cations is a quite difficult task resulting in general in a only partial exchanged zeolite. This fact has to be taken into account when comparison between different cations is considered. Another important information present in this table is the surface area of the materials that provides indication about the presence of defects in the structure, in particular about structure collapse due to improper synthetic or activation procedure. For what concerns Mg-exchanged zeolites, the activation temperature has another important effect: it is well known that Mg ions introduced by exchange in aqueous solution are at least in part in the form (Mg–OH)+.203 High activation temperatures (about 400 °C) are then needed in order to completely dehydroxylate (Mg–OH)+ to form Mg2+.237
CO2 affinity dependence upon different cations has been studied in literature in four different zeolitic frameworks: KFI (LS-KFI and Mg-ZK-5),233,236 CHA,234 LTA76 and FAU (zeolite X)76 zeolites. In these studies, the capacities of Mg-exchanged zeolites were compared to the ones obtained for the corresponding materials exchanged with other alkali and alkali earth cations. By comparing the results reported, the effect of the framework topology on the cation reactivity resulted to be important. This is a very well known matrix effect of zeolites and it is particularly important for an ion as Mg2+, having a very high charge/radius ratio. For that reason, a common trend cannot be evidenced for the different frameworks.
Different trends were also obtained for materials having the same framework topology but different Si/Al ratios. In LS-KFI materials (low silica KFI), the Mg-exchanged material showed the lowest CO2 capacities in the 0–1 bar range than all the other LS-KFI frameworks considered (Li+, Na+, K+, Ca2+, Sr2+) likely because Mg2+ is more shielded than the other ions by the negative zeolite framework.236 Nevertheless, the low crystallinity of the Mg-KFI framework in this study is evident when comparing the surface areas for the different counterions, being for example Ca-KFI possessing a larger surface than Mg-KFI, otherwise than expected (337 vs. 358 m2 g−1, respectively).236 For what concerns separation, LS-Mg-KFI showed the larger selectivity at 308 K and 1 bar in a 50% CO2/CH4 flow (62), that is identical to NaX.236 The corresponding high silica material, Mg-ZK-5 showed the lower uptake for p < 0.3 bar with respect to other alkali and earth-alkali cations, ascribable also in this case to the low accessibility of the Mg2+ cations.233 This was confirmed by the low qiso obtained also at the lowest CO2 coverage (see Table 7). Nevertheless, at 1 bar an uptake comparable to NaY and Li-ZK-5 was obtained.233 Interestingly, the shape of Mg-ZK-5 isotherm, being less steep than Li-ZK-5 ones, indicates that larger working capacity would be possible by using this zeolite in PSA systems without the need of the more costly VSA systems, as for the other zeolites under study.233 For CO2/N2 flow of 10/90 in a PSA system working between 5 and 1 bar, Mg-ZK-5 resulted to have a decidedly higher selectivity with respect to 13X (121 vs. 37) and higher working capacities (2.05 vs. 1.44).233 Its higher working capacity is related to the lower CO2 isosteric heat in Mg-ZK-5 with respect to 13X.233
In Mg-CHA zeolites, on the contrary, being the Mg ion more exposed, higher isosteric heat was obtained with respect to alkali and earth alkaline analogues, with corresponding poor performances as material for CO2/N2 separation.234 Energetic of interaction reported for LTA and FAU frameworks followed the same trend than in KFI framework, although likely slightly biased by a too low activation temperature of 250 °C.76 The reason of the low CO2 isosteric heat in MgA has been explained on neutron diffraction basis and shows as the so called “framework shielding effect on the cation” can be more complicated than in general imagined.76
Neutron diffraction studies evidenced as the Na+ cations occupies 6-ring and 8-ring in the LTA framework.238 A study conducted on CO2 loaded LTA zeolites76 evidenced as CO2 has two preferential adsorption sites (see Fig. 16). In site A, CO2 interacts with two cations at the same time through the oxygen atoms.76 On the contrary, in site B the carbon atom sites in the 8-ring plane, if free, to CO2 contemporaneously interact with two oxygen atoms of the framework.76 Between the two sites, site B is the most energetic having a double occupancy with respect to A.76 This site would be at the basis of the particularly high isosteric heat registered for CaA (58 kJ mol−1) with respect to other alkali and alkali earth exchanged LTA.76 For what concerns MgA, the unexpected lower qiso measured with respect to Ca-A were explained by a partial blocking of 8-ring sites by Na+ due to a lower degree of exchange with respect to Ca-A or to a lower exposure of Mg2+.76 Nevertheless, another explanation can rely in the low activation temperature adopted that would not allow a complete dehydroxylation of the (Mg–OH)+ species.
Metal organic frameworks are often seen as an evolution of zeolites, being MOFs in principle applicable to all the uses already covered by zeolites, with the advantage of the higher structure design flexibility, deeply described in Section 4.3. Nevertheless, at present, MOFs are in general seen as a laboratory curiosity more than materials that could have a real practical use. In fact at present MOFs cannot compete with zeolites because of the larger thermal, pressure and chemical stability and lower cost of zeolites. CO2 separation from atmosphere and flue gases, nevertheless, is expected to be one field of exception.10 With respect to zeolites, MOFs have particular advantages, being CO2 adsorption less affected by water than in zeolites129,239 and being water more easily removed from MOFs than from zeolites.129 For example IRMOF-74-I can be completely dehydrated for treatment at 200 °C,144 whereas for zeolites such a condition is reached only for temperatures of about 350–500 °C.240,241 MOFs moisture sensitivity, nevertheless, is a strong disadvantage.
Amine grafting in zeolites has been proved to be a good strategy in order to enhance the water resistance of zeolites as CO2 sorbents in CO2/CH4 separation, although a general lowering of the CO2 capacities is contemporaneously observed.10 This observation is analogue to what reported only recently for MOFs (see Section 4.3). A new synthetic method242 where the amine molecules (TEPA = tetraethylenepentamine) were introduced in zeolites during the synthesis seems to be particularly promising. In this case, an increase in the CO2 uptake was observed. Nevertheless, the high environmental impact of amines indicates that them do not represent a possible solution on the basis of the large scale of sorbents needed to sequestrate CO2.63 Clusters of MgO have been introduced in Y zeolites by heating Mg-exchanged Y zeolites,203 through their impregnation with magnesium dimethoxide203,243 or by direct dispersion by microwave.201 Strong basic sites are obtained only if ensembles of Mg and O form an MgO lattice.203,243 In this case, carbonates are formed very easily even from atmospheric CO2.203 The presence of oxide clusters enhances the amount of CO2 adsorbed and the zeolite toward CO2, allowing the atmospheric CO2 sorption at RT.203 This observation is particularly important because these clusters could represent a possible alternative to amines. Some investigations on this topic are undergoing in our laboratory, showing actually as the presence of the MgO particles not only increases the water stability of the adsorbent but an increase of the CO2 uptakes has been also observed.244
Zeolites have been also shown to be suitable supports for Mg(OH)2 nanoparticles, allowing to enhance their CO2 capacities up to the stoichiometric capacity (see Section 4.7.2).217
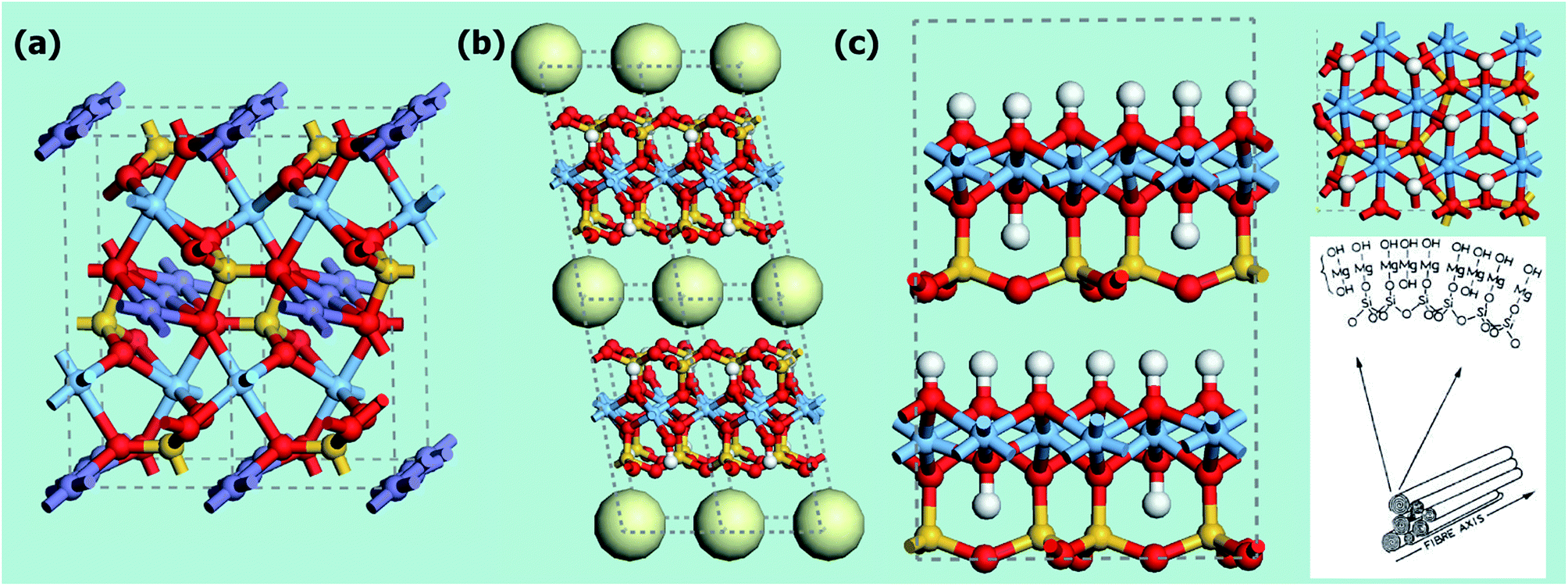 | ||
| Fig. 17 (a) Structure of olivine FeMgSiO4 from ref. 257. The atoms are represented according to following color code: hydrogen (white), oxygen (red), magnesium (light blue), silicon (yellow), iron (violet). (b) Structure of Cs-montmorillonite (Cs0.62[Al3.01Fe(III)0.41Fe(II)0.04Mg0.54](Si7.8Al0.2)O20(OH)4) from ref. 258. Tetrahedral atoms (silicon and aluminium) are represented as yellow sphere, whereas octahedral atoms (aluminium, iron and magnesium) are represented by light blue spheres. For the other elements, the following color code was used: hydrogen (white), oxygen (red), cesium (light yellow). (c) Chrysotile mineral description. From left to right: side and top view with respect to the brucite-like layers of the crystal ideal structure of chrysotile [Mg3Si2O5(OH)4]; schematic representation of chrysotile asbestos fibres (Reproduced with permission from ref. 259. Copyright 1999 Royal Society of Chemistry). The same color code as in part (a) was adopted. | ||
Clays are naturally abundant materials246 and for this reason they have attracted interest as CO2 sorbents. Nevertheless, clays are in general characterized by medium-low surface area at difference of what expected (<100 m2 g−1).247,248 This is due to the inaccessibility of the interlayer space to molecules in the gas phase, at difference of what happens in liquids. For this reason, the CO2 uptake of natural occurring clays are very low. In order to increase the surface area, a common strategy in layered materials science is to introduce species of appropriate size in the interlayer space (see also Section 4.7.2). The intercalation of those species can be obtained during the synthesis or by post-synthetic methods, the simplest of which is the ion exchange. In this way, the pore dimensions can be tuned up to the exfoliation of the material for very large species. Several solutions were proposed (aminopropyltrimethoxysilane, polyglycerol dendrimers, polyethylenimine, TEPA, …).63 Among them, amines resulted to be the most suitable intercalated species for clays to be used as CO2 scrubbers.63 It is interesting to notice that the introduction of TEPA, analogously to what verified for zeolites and MOFs, allowed to strongly increase the CO2 capacities of bentonite (a natural occurring material, mainly composed by montmorillonite clay, see Fig. 17b). CO2 capacities were increased from the negligible value reported for raw bentonite to up to 3.0 mmol g−1 at 75 °C and 1 bar.249 Also in this work, as for IRMOF-74-I, a bell bottomed dependence of the CO2 capacity on the amine loading was observed,249 indicating as the amount of the harmful and costly amines have to be always optimized.
| (Mg,Ca)xSiyOx+2y+zH2z + xCO2 → x(Mg,Ca)CO3 + ySiO2 + zH2O + heat |
Carbonation of silicates is an exothermic reaction (see Table 1), although the heat released during carbonation is lower than MgO/CaO oxides.60 Because of that, carbonation in magnesium silicate is a spontaneous process and, in fact, natural weathering is able to cause the carbonation of the fine-grained material on the surface of tailings pile with the formation of stable carbonate as magnesite (MgCO3), hydromagnesite (Mg5(CO3)4(OH)2·4H2O)) and pyroaurite (Mg6Fe2(CO3)(OH)16·4H2O) at atmospheric temperature and pressure conditions.254,255
It has been shown as in presence of mafic/ultramafic rocks (basic rocks, with a high Mg and Fe content), fixation of 1330 g C m−2 per year can be also reached, through natural weathering.255 Nevertheless, for what concerns bulky particles, instead, pretreating of the materials and high temperatures and pressures of reaction (>500 °C, >50 bar) are needed in order to obtain significant carbonation degrees, compromising the CO2 efficiency and the cost of the whole process. The reaction is in fact characterized by slow reaction kinetics because of the low surface area and the rapid formation of passivative carbonate or silica layers. Different routes for MCT have been then suggested, that can be divided in three main groups:60 (i) ex situ MCT; (ii) in situ MCT; (iii) other MCT (e.g. natural processes as passive MCT and biomineralization). Ex situ MCT refers to the aboveground carbonation of natural minerals and industrial alkaline wastes with (indirect MCT) or without (direct MCT) previous extraction of Ca or Mg. The extraction procedure is aimed to the separation of iron and to the elimination of the siliceous part, that is inactive in the reaction and moreover brings to the formation of a passivating layer. The alkaline element is then extracted as oxide or hydroxide and used in the carbonation reaction. The reaction kinetics in indirect MCT are decidedly larger than in direct processes in milder conditions.60 Moreover, a pure carbonate is obtained as end-product. In situ MCT processes have been only recently developed and consist in geological storage conducted in optimized conditions (surface area, temperature, pH and CO2 pressure) to accelerate the natural process.60 In situ processes are cheaper than ex situ processes although still higher in cost with respect to geological storage in sedimentary basins and characterized by the same environmental problems. It is also important to remind that the amount of carbonate produced would be so high (see Fig. 2c) that in order to lower the cost of production, applications of these products have to be addressed60 or other motivations of the carbonation process, besides CO2 capture itself, have to be envisaged.
Chrysotile. Chrysotile (Mg3Si2O5(OH)4), the asbestiform polymorph of serpentine, is a phyllosilicate constituted by tetrahedral [SiO4]4− sheets (T) and trioctahedral brucite-like [Mg3O2(OH)4]2− sheets (O) (see Fig. 17c). Each T layer is chemically bounded to an O layer through the sharing of oxygens between Mg and Si. The TO couples are bounded to the others through hydrogen bonding between the hydroxyls of the O layer of one TO unit and the oxygen of the T sheet of another TO unit. The hydroxyls in the brucite-like layer are of two kinds: Si–O–Mg–OH and OH–Mg–OH. These species are alternating at the nanotube surface, as shown in the scheme reported in Fig. 17c. As it is evident in this picture, one of the two hydroxyls of the OH–Mg–OH unit points towards the inner part of the TO sheet. The TO sheet lateral misfit is accommodated to a degree by a change in curvature resulting in various curved crystal shapes and sizes. The resulting material is composed by hollow nanotubes with an average diameter of 400 Å and inner diameter of 35 Å.254 As can be inferred from its tubular shape and composition, this mineral is an asbestos, that are well known carcinogenic agents. It is interesting to know, for example as chrysotile mining in the past has bring to the accumulation in Thetford Mines and Asbestos (Québec, Canada) of 2 × 109 tons of waste, rich in chrysotile.256 Similar sites are present for example in Australia,255 Italy,260 United States of America and Brazil. The importance of reclaiming of the land by removing chrysotile is then evident. The presence of a brucite-like surface is expected to bring a strong ability to react with CO2 to chrysotile. Nevertheless, in front of a 1
:1 CO2 to Mg molar ratio for the total conversion of chrysotile to hydromagnesite, only 0.01 was reached at about 130 °C (CO2 pressure of 31 bar, 1 bar H2O) and 0.15 at 375 °C (H2O/CO2/He = 10/90/50 ml min−1). This was explained by the low surface area of the material (20 m2 g−1 for the raw chrysotile)254 and by an even closer atomic distances in the O layer with respect to brucite that would not allow an easy diffusion to CO2 during the reaction. Alkali metal doping (10 wt%) was found to cause the increase in CO2 fixation up to a factor of 3.254 Further improvements were obtained by the observation that the presence of water is beneficial on carbonation, likely because of the formation of meta-chrysotyle (Mg3Si2O7),256 characterized by an amorphous structure with tendency to crystallize in the less harmful forsterite (Mg2SiO4).261 In particular, raw chrysotile was pretreated up to 700 °C (100 °C min−1, isotherm for 20 min) to form meta-chrysotyle256 before to be exposed to CO2. At difference of the pristine chrysotile, this material was able to strongly react with CO2 in presence of steam for temperature between 100 and 160 °C, with a peak of 0.7 mol CO2/mol Mg at about 130 °C (CO2 pressure of 31 bar, 1 bar H2O). This is a strong enhancement with respect to pristine chrysotile in the same conditions (1 mol%) and was observed only when water is present in the reaction vessel. XRD analysis indicated the formation of magnesite, while X-ray photoelectron spectra revealed the formation of both carbonate and bicarbonate species.256 The formation of these species was found to not be related to the presence of brucite or other impurities. This enhanced activity was explained by a rehydroxylation in steam accompanied by a strong increase in the surface area (13 and 147 m2 g−1 for meta chrysotile not exposed and exposed to steam, respectively). The formed species resulted to be highly stable, being decomposed only in the 400–600 °C range.256
4.9. Hydrides
Hydrides are well-known reducing agent used in many chemical reactions for this purposes. As discussed in Section 2.2.1, hydrogenation of CO2 would represent an advantageous method to obtain fuels having lower, if not negative, carbon footprints. In this respect, hydrides have been employed as (i) catalysts35 in the CO2 hydrogenation or as pure35,262 or as supported species.14,71 Moreover, their use as (ii) as co-reactants21,263–265 has been also reported.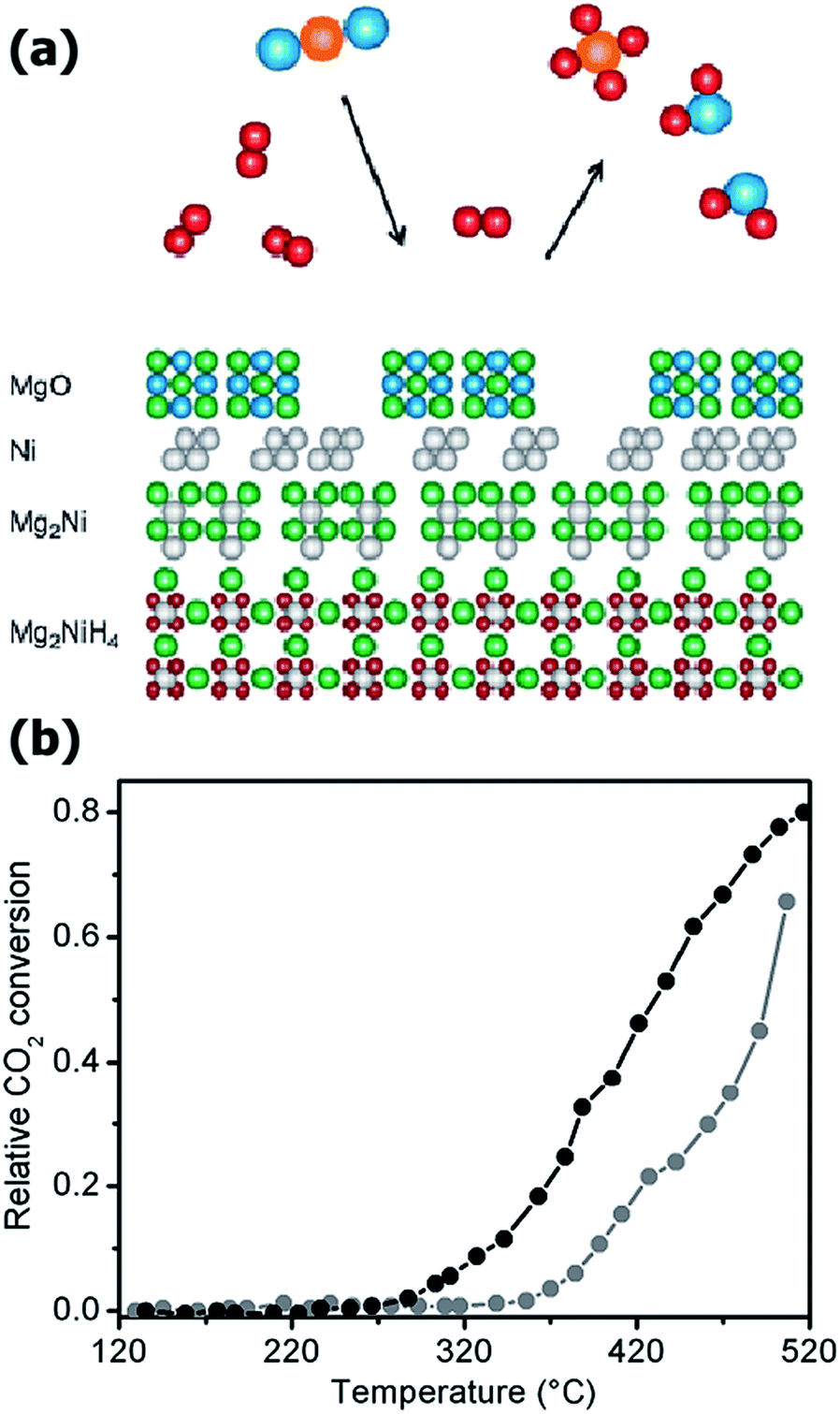 | ||
| Fig. 18 (a) The disproportionated surface of Mg2NiH4 (schematic representation). The metal hydride surface becomes catalytically active as Ni precipitates and converts CO2 into CH4 during hydrogen desorption (CO2 + 4H2 CH4 + 2H2O). Reproduced with permission from ref. 35. Copyright 2012 Royal Society of Chemistry. (b) The relative CO2 conversion as function of the temperature (5 °C min−1) for CO2 hydrogenation in a flow of CO2 and H2 (H2/CO2 = 8 with 50 ml min−1 He at a total flow rate of 100 ml min−1) on Mg2NiH4 for the first cycle (grey scatters) and after 18 cycles (black scatters). Data digitalized from ref. 35. | ||
A closer glance to the disproportionate surface of Mg2NiH4 (Fig. 18a) shows its essentially coincidence on the chemical point of view with the case of metal particles supported on metal oxide. It is interesting to notice as, in fact, only intermetallic hydrides constituted by a R component, which the correspondent oxide is able to allow an easy formation of carbonates, are active as catalysts in CO2 hydrogenation. This is due to the fact that35 in the CO2 hydrogenation reaction, the dissociation of the H2 molecule is not the rate controlling step but the dissociative sorption of CO2 molecule on the hydride surface.35 The same results was reported for oxide catalyzed hydrogenation reactions. This coincidence could allow interesting perspective in the design of new optimized catalysts for CO2 hydrogenation. Results and conclusions driven for one class of systems can be then applied to the other. This fact is in general not underlined in the literature on the subject. For example, the evidence that the rate controlling step in CO2 methanation is the CO2 dissociation is self-evident for hydride systems. Moreover, strategies for catalyst protections over poisoning could be straightforwardly driven from disproportioned hydride surfaces to supported metal particle systems. Use of this catalyst for dry methane reforming is expected to bring some improvements in the stability of Ni based catalysts especially for what concerns coke formation.
Porous complex hydrides are a new class of materials in the panorama of metal hydrides.284 Among them, gamma phase of Mg(BH4)2 possesses the interesting peculiarity to show a large permanent porosity, accounting for about the 33% of the material volume (see Fig. 19).284 Its structure is constituted by Mg2+ ions in tetrahedral coordination, linked through a shared [BH4]−, giving rise to a highly 3D porous structure characterized by hexagonal overtures with a narrowest dimension of 5.95 Å (geometrical distance). Each hexagon, having a chair conformation, shares every Mg–BH4–Mg side with a vicinal hexagon through a tetrahedral angle. The high surface of this material (1160 m2 g−1) makes it interesting as nanosponge for gas adsorption and its ability to adsorb large quantities of molecular hydrogen was recently reported.284 This material, at difference of all the previously reported hydrides, is able to react fast with CO2 at RT and for pressure close to the atmospheric CO2 partial pressure. In Fig. 19, the CO2 reacted as function of time at 30 °C and 1 bar is reported (blue curve), showing as 1 CO2 reacts every 11 [BH4]− after only 11 min. The plateau value of 12 mol kg−1 CO2 was observed only after 7 days. Key role of the surface area was confirmed by comparison with data collected on its unporous isomorph α-Mg(BH4)2 (Fig. 19, dark cyan line). The reaction products were analyzed by 13C-NMR and IR spectroscopies indicating as formate, methoxy and alkoxide species are formed, most of them bound to the boron atom. The reaction was monitored in time by IR spectroscopy, evidencing as all the products are formed at once (in the time needed for the spectra recording) and no evolution of the products towards more reduced CO2 species was observed, at difference of ammonia borane studies.275,276,278,279,283 No evolution of gaseous reaction species (e.g. methane or CO) was also evidenced at 30 °C and 1 bar.21 On the contrary, exposure to supercritical CO2 (40 °C and 90 bar for 1 h)263 allowed the formation of methane and methanol, further underlying the importance of the reaction conditions on the products that can be obtained in hydrogenation reactions of CO2.
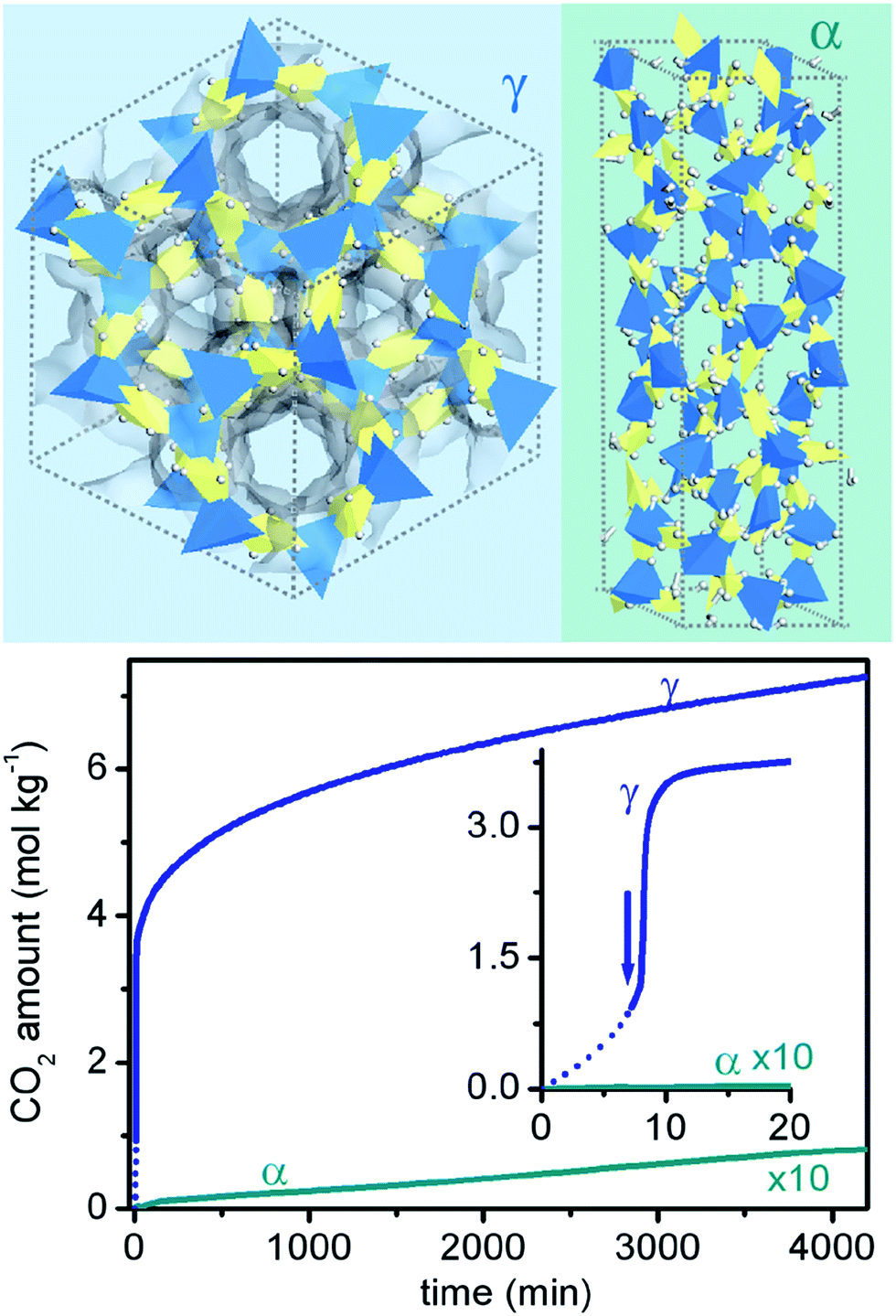 | ||
| Fig. 19 γ-Mg(BH4)2 and α-Mg(BH4)2 structures: Mg atoms are represented as light blue tetrahedra, B atoms as light yellow polyhedra and H atoms as spheres. The Connolly surface on γ-Mg(BH4)2 structure obtained by using a probe with radius of 1.82 Å is represented. (a) Kinetic curve of CO2 reaction with γ-Mg(BH4)2 at 30 °C and 1 bar (blue curve). The inset shows the first 20 min of reaction (the arrow indicates the end of the CO2 dosing procedure). The analogous curve obtained for α-Mg(BH4)2 is reported for comparison (dark cyan curve): the astonishing effect of the increased surface area on the reaction time is well evident. Data from ref. 21. | ||
An important drawback of the use of hydrides as reactants is the impossibility to rehydrogenate them after reaction with CO2 (ref. 21, 263 and 265) and γ-Mg(BH4)2 does not represent an exception, likely because of the higher CO2 interaction energy with respect to H2 and the large stability of the B–O bonds.21 If re-hydrogenable, γ-Mg(BH4)2 could be used as catalyst instead as reagent with performances in temperature and pressure conditions close to atmosphere not achieved by any other system. Nevertheless, this system remains very interesting. In fact, the very fast kinetics associated to the reaction provide to this hydride the peculiar characteristics to be able to couple in one step the separation and recycling of CO2 in a wide temperature (RT < T < 190 °C) and pressure range allowing to avoid the storage and transportation steps.21 The possibility to use γ-Mg(BH4)2 as co-reagents in other CO2-based reactions to enlarge the number of obtainable products has been also proposed.21
5. Conclusions
In the present review, many of the different systems at work in both the natural and the artificial CO2 cycle were described by using as leitmotif the presence of Mg in their structure. Although breakthrough results were reported recently for all the steps of the synthetic CO2 cycle,52,57,147,151 there is still large room for improvements.For what concerns the catalysis research, among all the possible reactions involving CO2, the synthesis of fuels has a principle role. This is appealing not only because it reduces the carbon footprint of fuels but also because it would allow to maintain the present existing infrastructure and the same quality of life, being most of the energetic alternatives to fossil fuels far to allow the same performances. In some applications (as plane propulsion), at present a reasonable substituent does not even exist.285 Considering the fact that 73.8% of the CO2 emissions are related to fossil fuels burning, it is clear that any strategy for CO2 reduction will need to address also to: research on CCS and CCU, energy efficiency, and the use of less carbon intensive fuels such as CH4 and other renewable energy sources.286 Coal-fired plants for example are characterized by a very low energetic efficiency (30%)55 and they represent the most important CO2 stationary sources. Their substitution has to envisaged by new coal-fired plants characterized by a more careful heat management55 or by plants based on more efficient processes such as oxyfuel combustion coupled with CCS. Such changes can be addressed effectively only at the political level.
Coming back to CO2 recycling, personally I consider the carboxylation of alcohols to fuels and the dry methane reforming as the most important reactions. The latter in particular, in combination with the Fisher-Tropsch process, has the advantage to transform the two most important greenhouse gases (CO2 and CH4) into hydrocarbons.28 This technology is particularly promising for CO2/CH4 mixture deriving from fermentation and it does not require for CO2 separation. In this field, the fast deactivation of industrial catalysts due to coke formation is the most important problem to overcome. A broader use of materials with high oxygen mobility such as perovskites is expected to bring benefits from this point of view.28,40 Particularly interesting would be the testing of natural siliceous perovskites as (Mg,Fe)2SiO3, Al-(Mg,Fe)2SiO3. Carboxylation of methane, although more rarely studied, is another appealing reaction for valorization of biogas.12
Nevertheless, the availability of efficient catalytic processes is not enough to make CO2 utilization feasible. In fact, the high cost of CO2 separation makes it a costly feedstock, a fact that is at the basis of its low exploitation as a reagent in the industry.15 The drivers that can allow to reverse this situation are resources scarcity on one hand but also political and regulatory actions. The Kyoto protocol restrictions on CO2 emissions can for example create a market for captured CO2, aimed to compensate in part the cost of separation. On the other hand, reactions that allow to avoid the separation step have to be sought, like for example dry methane reforming and CO2 hydrogenation by hydrides.21,35,263
For what concerns separation, this is the field where improvements would be more urgently required, firstly to lower the energetic cost of CO2 separation for the reasons discussed above and moreover to improve the purity of separated CO2 (indispensable in many reactions).15 In this field the necessity of new materials is particularly urgent.63 Among the different materials reported for low temperature separation, MOFs, because of their large flexibility in design, are particularly promising for improvements. The possibility to chemically alter the interior of MOFs to create pores analog to the enzyme pockets that can match target molecules is very promising.119,287 Flexible structures of MOFs able to respond to external stimuli of different nature (pressure, specific adsorbates, light, …) have been also reported,81,133,287–289 materials that can bring some breakthrough in the separation field. Although the large selectivities and working capacities reported for some MOFs are very high (see Table 5), this class of materials suffers of two main problems: (i) high cost; (ii) water sensitivity. The low water resistance of MOFs does not only lower their performances as CO2 scrubbers in presence of moisture as observed for zeolites, but it undermines the stability of the materials themselves, besides some important exceptions.130,153,289–291 MOFs composites involving the presence of carbon, graphene oxide, aminated graphene oxide, nanotubes, etc., because of their hydrophobic nature, showed better anti-moisture performances than pure materials.292–294 Other strategies are represented by the introduction of hydrophobic groups, such as –CF3.295 The best performances in CO2 separation were observed for materials containing grafted alkylamines, in analogy to what reported for almost all the other classes of materials.63,130,147,148,152,296 However, the amount of sorbents needed to address global scale CO2 capture has to be so high that the choice of environmental benign compounds is mandatory.63 Amines-based materials are lacking in this respect being corrosive and carcinogenic compounds.
MOFs are also materials characterized by a very high cost. Nevertheless, the cost of the material is in many cases only a small fraction of the cost of the CO2 separation process, whose main contribution is energy consumption. Moreover, economical cost of CO2 separation, although important, has to be compared with the corresponding cost due to climate change effects (e.g. drastic weather events, loss of fertile land and the reduction of Earth land surface due to sea level rising). Low cost materials for CO2 capture exist and they are almost exclusively carbon based materials obtained from waste resources.63,297 This solution has a double benefits allowing to reduce the waste (and also the biogas evolved from its fermentation) by creating an effective CO2 sorbent that can be produced in the huge necessary amounts.200 In fact, 140 Gtons per year of biomass are produced from agriculture.200 These carbons are often nitrogen rich or contain nanodispersed basic oxides which improve their CO2 sorption capacities.63 Carbons obtained from poplar anthers are able to adsorb up to 51.3 mmol g−1 at 25 °C and 50 bar.298 Although carbons are good storage materials, they are in general characterized by very poor performances in separation processes.63 An exception is represented by the MgO/C system reported in ref. 200 able to capture significant CO2 quantities also at 1 bar. Moreover this system was obtained by fast pyrolysis of biomass impregnated with sea salts.
Among separation processes, separation from the atmosphere is the one needing more improvements. Unfortunately, this is also the process for which the improvements are more urgent, being the drastic effects of the abnormal rise in CO2 concentration in the atmosphere already visible and asking for an action that cannot be delayed further. Actually, few materials have been reported to date having significant CO2 capacities at the very low partial CO2 pressures at play (0.4 mbar). The best performing materials reported up to now are all amine containing structures (see for example Table 5). Nevertheless, the presence of amines still makes them unsuitable for a global use and production at least on the Mtons scale. The short list of materials can be enlarged by considering for example the possibility to compress air before capture. Normal compressors allow to reach pressures up to 35 bar. This would correspond to an increase in the CO2 partial pressure up to 14 mbar, enabling the use of a broader range of existing capture materials. An increase of CO2 partial pressure could be also reached through a two step separation process where the purity of separated CO2 is increased stepwise. Being the amount of CO2 to be handled so large, in order to be effective, an organized action at planetary level would have to consider no more than 2–3 ways to capture CO2 from the atmosphere. In the choice, the energy and time needed to build the CCS infrastructure have to be considered. In the end, at present any synthetic material proposed so far does not constitute a reliable choice for CO2 capture from the atmosphere.
For what concerns natural processes, two have shown to be able to allow significant carbon fixation from the atmosphere: natural weathering of silicates (Section 4.8.3) and photosynthesis (Section 4.2). In Table 8, a comparison between these two processes and a membrane capture plant based on one of the best performing material reported for CO2 capture (IRMOF-74-IIb-mmem) is reported. The analysis presented there is far to be quantitative being based on rough assumptions. These assumptions have been adopted purposedly in order to put on the same plane such different methods to roughly estimate the order of magnitude at play in term of time and cost. In fact, the estimate of times, precise or rough in nature, are completely lacking in the literature. For what concerns the photosynthetic and mineralization processes, data on carbon fixation are often normalized to the surface area occupied by the system. In order to compare MOFs with them, it has been considered the amount of IRMOF-74-IIb-mmen that would be necessary to cover with a thickness of 1 cm a certain surface. As surface, the total land surface has been considered. Although this option is far from being feasible, it can be though as an upper limit. Among the several plantae existing, hybrid poplars have been considered here. In fact, they rank second among the fastest growing plants on Earth and they are not as strongly invasive and eco-system killers as the bamboo. Moreover, its wood is considered a medium high quality wood for combustion.302 For what concerns mineralization, on the contrary, the time for the completion of the process has been calculated considering the two limit carbon fixation rates reported for natural weathering in ref. 255.
| Process | Product | Volume occupieda (EB units) | Time to complete the separationb (year) | Characteristic storage timec (year) | Costd (USD2010 per ton CO2) | Air compression/intake |
|---|---|---|---|---|---|---|
| a Volume normalized to the volume occupied by 900 metric Gton of liquid CO2 to facilitate the comparison (see Fig. 2).b Comparative time calculated in the extreme hypothesis to completely cover a surface equal to the Earth land surface with: (i) 1 cm thick layer of IRMOF-74-IIb-mmen (for liquid CO2), (ii) mine tailings able to provide an average capture of 27–1330 g C m−2 per year.255 and (iii) an hybrid poplar wood.299 For the MOF, the time necessary for the sorption/desorption cycle was underestimated and considered coincident twice the time needed for saturation of the material in 15% CO2 atmosphere (15 × 2 = 30 min).147 For the hybrid poplar wood, the growth factor was calculated from the values reported in ref. 300. See ESI for further details.c Values from ref. 55 and 299.d Cost of the separation process with MOFs by considering it coincident with those reported for membrane post-combustion capture processes in ref. 64. The cost of the material production (considering an operational time of the plant larger than 1 year and a market cost of 1 % kg−1 for the scrubber) resulted negligible with respect to the separation process itself in the approximations adopted in this work (<1 % ton per CO2). The cost related to the air intake process have not been considered, although expected to be particularly important in air sequestration. Moreover, at present such an infrastructure does not exist and then the cost of this process could be only of speculative nature. Mineral carbonation cost from ref. 229. Upper limit for carbon fixation in plant estimated as the cost for wood production from ref. 301. | ||||||
| Separation through MOFs | Liquid CO2 |  |
0.4 | ? | 46–90 | Yes |
| Mineral carbonation | Carbonate |  |
1.24–61 | >105 | 0–8 | Yes/no |
| Photosynthesis | Wood |  |
6–39 | 10 to 102, >105 | 0–120 | No |
Surprisingly, the time estimated to complete the separation (in the rough approximation reported here) covers a few orders of magnitude. Actually, among the values in Table 8, the one reported for the artificial process is particularly low. Longer periods, although of only one order of magnitude larger, would be necessary to photosynthesis to fix the same amount of CO2. For what concerns the separation cost, mineral carbonation remains the cheapest solution, having the photosynthetic and the MOF comparable cost. In the calculation of the data reported in Table 8, it is important to note that the time needed to construct the infrastructure necessary for each process has not been considered. This approximation is not very important for the two natural processes, being only an extension on the global scale of their already large exploitation. On the contrary, this is expected to strongly delaying the artificial process to start. The inertia in introducing new technologies is a well-known problem in this subject and their implementation can be driven only by policy makers or financial incentives.63 Moreover the synthesis of the huge amount of sorbent necessary is not a trivial industrial pursuit.
Another important point to consider in this approximate comparison is the end product of the three separation processes. For what concerns the artificial process, pure CO2 is obtained, which represents an interesting reagent and solvent, as discussed along this review. Nevertheless, besides the small part exploited for this aim by industries, the most part of this amount would need to be stored in a long term disposal site (mineralization) increasing the cost and complexity of the process. Mineralization and photosynthetic end products represent themselves a solution for the long term storage of CO2. Inorganic carbonates have the advantage to represent the ideal CO2 disposal form, because of their high stability. Moreover, carbonates are considered valuable products, with uses in different fields from cosmetics, pharmaceutics industry, paints, ceramics and cements. Magnesium bicarbonate is an important additive for desalinated and mineral water. A value of silicate carbonation would be added if this reaction would be used for land reclamation, as shown in the case of chrysotile (Section 4.8.3). On the other hand, for what concerns photosynthesis, a stable storage form is wood whose durability and inertness enable carbon storage over a relatively large timescale, comparable with that of carbonates.299 For this reason, it has been previously suggested that forestation can be an effective way to reduce global warming.299
Moreover, wood represents, among the three end products reported in Table 8, the one with the highest added value coupled with a significant chemical stability.299 By hypothesizing a correct exploitation of trees as carbon fixation systems (substitution of the trees each 3–5 years, appropriate storage conditions of wood, etc.), the use for this purpose of a surface corresponding to only 10% of the Earth land surface would allow to observe in 2050 the same atmospheric CO2 concentration measured today. If combined with proper carbon sequestration at the larger stationary sources, in this optimistic view a deflection of the trend could be observed. Nevertheless, the forestation solution presents important drawbacks, essentially related to the necessity to exploit the same resources of agriculture: water and land surface. Actually, the first reason at the basis of deforestation is the conversion of forests in arable lands, because of the increasing food request. This means that such an action could have a direct and important effect on the price of food. For what concerns soil occupations, solutions not requiring the reconversion of arable lands to forests have to be considered. Possible solutions can be represented by a larger introduction of trees in urban areas or the use of reforestation as a mean for land reclamation of polluted arable lands. For what concerns the massive use of water, the optimization of the use of water in agriculture is at present largely studied. Large amounts of freshwater are commonly used also in material synthesis and CO2 underground sequestration processes. A comparison of the three processes reported in Table 8 by LCA, comprehensive of these aspects, is at present not possible on the basis of the literature data. It would be particularly interesting if it would be addressed in future studies.
The most important advantage of photosynthesis is that, as also stated by Lackner,55 seems currently the only practical form of air capture in the short middle term. It is interesting to note in this regard that this would have a precedent in the Earth's history. In fact, a particular algae species, the Azolla filliculoides, among the fastest growing plants on Earth,303 was responsible of the strong decrease in the CO2 level in the atmosphere about 48.5 million years ago, during the Eocene. This event goes under the name of the Azolla event.304 The massive proliferation of this freshwater fern in the Artic region up to 4 106 km2 of covered surface (due to the mild temperature, to the presence of a freshwater surnatant layer in the oceans, and to the low presence of microorganisms for degradation) caused the decrease in CO2 in the atmosphere from 3500 to 650 ppm in 800000 years.90,304 Several varieties of Azolla are still cultivated. Algae have in fact several uses as green manure, fodder for poultry,305 can be used as cosmetics, whole food ingredients, food integrators,306 for bulk chemicals, paper,307,308 bio-ethanol308,309 and biofuels production.306 Moreover, microalgae need for their cultivation only 3% of the area required by vegetation,306,310 representing then an interesting alternative for CO2 fixation. Nevertheless it is important to stress as the efficient CO2 sequestration during Eocene was possible because of the absence of microorganisms responsible for fermentation of biomasses that exist nowadays. At present, the biogas that would be generated by putrefaction of microalgae, if not correctly handled, would cause a strong worsening of the greenhouse effects instead to be beneficial. The problem of degradation is only marginal in wood production but nevertheless it has to be properly addressed because of the large amounts of leaves and other waste products that would be produced.
An important point that is often forgotten is that any cultivation causes an important production of volatile organic compounds and in particular of isoprene.311 Hybrid poplars are not an exception. Isoprene is known to react with NOx and radicals allowing a reduction of their concentration in the air.311,312 In the reaction, ozone is released causing the reduction of the concentration of aerosol particles and pollutants in the air.311−313 This observation is important because indicates that the introduction of trees would be particularly beneficial in high polluted urban areas. Nevertheless, isoprene has been determined to critically influence the planet climate and then, careful modeling of any increase in its production have to be made. Moreover, ozone is a priority air pollutant causing about 22000 excess deaths per year in Europe.311 For these reasons, all the possible parameters have to be carefully considered before a global action is started to address the problem of CO2 excess in the atmosphere.
Like synthetic processes, also the photosynthetic one can be improved in efficiency. In fact, as stated in Section 4.1, Rubisco is the less efficient enzymes on Earth. For this reason, research is done extensively on how to increase crop yields by switching C3 plants to C4 ones (especially rice)87,108,314 or on increasing the Rubisco efficiency.84,87,102 This research is in primis aimed to decrease the world hunger. Nevertheless, besides the ethical and environmental concerns, a change in the carbon fixation rate is known to correspond to an alteration of the food nutritional parameters. An increase in the carbon dioxide fixation rates due to the increase in the atmospheric CO2 concentration, for example, lower the quality of the wheat.94–96 This can be related to the fact that during photorespiration most of the nitrate/nitrogen assimilation happens, necessary for protein production.315 In this way, faster growing plants would constitute a lower quality food. Moreover, accordingly to FAO one third of global food is wasted, indicating as the world hunger problem could be solved by a more careful planned distribution of the food resources.
Nevertheless, this research can be beneficial to biofuels production and carbon fixation purposes.87,101,306,308 Moreover, a deeper knowledge of the mechanisms and structural modifications in enzymatic reactions involving CO2 can help in the design of new synthetic catalysts and sorbents.
It is interesting to remember as in many cultures there was the habit to plant a tree when a child was born. In China, an Empress tree has to be planted for any newborn girl. It is not surprising that this beautiful shrub is among the ten fastest growing plants on the planet.
Acknowledgements
Prof. Adriano Zecchina and Dr Gabriele Ricchiardi are acknowledged for useful discussions on the global warming problem.Notes and references
- J. R. Petit, J. Jouzel, D. Raynaud, N. I. Barkov, J. M. Barnola, I. Basile, M. Bender, J. Chappellaz, M. Davis, G. Delaygue, M. Delmotte, V. M. Kotlyakov, M. Legrand, V. Y. Lipenkov, C. Lorius, L. Pepin, C. Ritz, E. Saltzman and M. Stievenard, Nature, 1999, 399, 429–436 CrossRef CAS.
- http://www.esrl.noaa.gov/gmd/ccgg/trends/, 2015.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- M. Mikkelsen, M. Jorgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43–81 CAS.
- J. D. Shakun, P. U. Clark, F. He, S. A. Marcott, A. C. Mix, Z. Liu, B. Otto-Bliesner, A. Schmittner and E. Bard, Nature, 2012, 484, 49–54 CrossRef CAS PubMed.
- Jet Propulsion Laboratory http://sealevel.jpl.nasa.gov/, 2015.
- D. A. King, Science, 2004, 303, 176–177 CrossRef CAS PubMed.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef CAS PubMed.
- F. Klein and T. M. McCollom, Earth Planet. Sci. Lett., 2013, 379, 137–145 CrossRef CAS.
- Z. Zhang, Z.-Z. Yao, S. Xiang and B. Chen, Energy Environ. Sci., 2014, 7, 2868–2899 CAS.
- D. M. D'Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef PubMed.
- H. J. Freund and M. W. Roberts, Surf. Sci. Rep., 1996, 25, 225–273 CrossRef and references therein.
- E. A. Quadrelli, G. Centi, J. L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194–1215 CrossRef CAS PubMed.
- W. Wang, S. P. Wang, X. B. Ma and J. L. Gong, Chem. Soc. Rev., 2011, 40, 3703–3727 RSC.
- M. Aresta, in Activation of Small Molecules, Wiley-VCH Verlag GmbH & Co. KGaA, 2006, pp. 1–41 Search PubMed.
- A. Behr, Carbon Dioxide Activation by Metal Complexes, VCH Verlagsgesellschaft, Weinheim, 1988 Search PubMed.
- J. G. Vitillo, M. Savonnet, G. Ricchiardi and S. Bordiga, ChemSusChem, 2011, 4, 1281–1290 CrossRef CAS PubMed.
- J. M. Weber and H. Schneider, J. Chem. Phys., 2004, 120, 10056–10061 CrossRef CAS PubMed.
- R. Vaidhyanathan, S. S. Iremonger, G. K. H. Shimizu, P. G. Boyd, S. Alavi and T. K. Woo, Science, 2010, 330, 650–653 CrossRef CAS PubMed.
- X. Kong, E. Scott, W. Ding, J. A. Mason, J. R. Long and J. A. Reimer, J. Am. Chem. Soc., 2012, 134, 14341–14344 CrossRef CAS PubMed.
- J. G. Vitillo, E. Groppo, E. G. Bardaji, M. Baricco and S. Bordiga, Phys. Chem. Chem. Phys., 2014, 16, 22482–22486 RSC.
- G. Busca and V. Lorenzelli, Mater. Chem., 1982, 7, 89–126 CrossRef CAS.
- B. Bonelli, B. Civalleri, B. Fubini, P. Ugliengo, C. Otero Areán and E. Garrone, J. Phys. Chem. B, 2000, 104, 10978–10988 CrossRef CAS.
- M. Werner, S. Hariharan and M. Mazzotti, Phys. Chem. Chem. Phys., 2014, 16, 24978–24993 RSC.
- K. Teramura, T. Tanaka, H. Ishikawa, Y. Kohno and T. Funabiki, J. Phys. Chem. B, 2003, 108, 346–354 CrossRef.
- M. M. Halmann, Chemical Fixation of Carbon Dioxide: Methods of Recycling CO2 into Useful Products, CRC Press, Boca Raton, Florida, 1993 Search PubMed.
- G. A. Olah and Á. Molnár, Hydrocarbon Chemistry, John Wiley & Sons, 2003 Search PubMed.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- Q. Cai, B. Lu, L. Guo and Y. Shan, Catal. Commun., 2009, 10, 605–609 CrossRef CAS.
- R. V. Siriwardane and R. W. Stevens, Ind. Eng. Chem. Res., 2009, 48, 2135–2141 CrossRef CAS.
- K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce Jr and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- J.-N. Park and E. W. McFarland, J. Catal., 2009, 266, 92–97 CrossRef CAS.
- J. Mascetti, F. Galan and I. Pàpai, Coord. Chem. Rev., 1999, 190–192, 557–576 CrossRef CAS.
- D. E. Peebles, D. W. Goodman and J. M. White, J. Phys. Chem., 1983, 87, 4378–4387 CrossRef CAS.
- S. Kato, A. Borgschulte, D. Ferri, M. Bielmann, J.-C. Crivello, D. Wiedenmann, M. Parlinska-Wojtan, P. Rossbach, Y. Lu, A. Remhof and A. Zuttel, Phys. Chem. Chem. Phys., 2012, 14, 5518–5526 RSC.
- S. Yokota, K. Okumura and M. Niwa, Catal. Lett., 2002, 84, 131–134 CrossRef CAS.
- J. R. Rostrup-Nielsen, K. Pedersen and J. Sehested, Appl. Catal., A, 2007, 330, 134–138 CrossRef CAS.
- G. Du, S. Lim, Y. Yang, C. Wang, L. Pfefferle and G. L. Haller, J. Catal., 2007, 249, 370–379 CrossRef CAS.
- P. J. Lunde and F. L. Kester, Ind. Eng. Chem. Process Des. Dev., 1974, 13, 27–33 CAS.
- H. Zhan, F. Li, P. Gao, N. Zhao, F. Xiao, W. Wei, L. Zhong and Y. Sun, J. Power Sources, 2014, 251, 113–121 CrossRef CAS.
- D. Scarano, S. Bertarione, F. Cesano, J. G. Vitillo and A. Zecchina, Catal. Today, 2006, 116, 433–438 CrossRef CAS.
- A. Parmaliana, F. Arena, F. Frusteri, S. Coluccia, L. Marchese, G. Martra and A. L. Chuvilin, J. Catal., 1993, 141, 34–47 CrossRef CAS.
- B. Nematollahi, M. Rezaei, E. N. Lay and M. Khajenoori, J. Nat. Gas Chem., 2012, 21, 694–702 CrossRef CAS.
- D. Pakhare, C. Shaw, D. Haynes, D. Shekhawat and J. Spivey, J. CO2 Util., 2013, 1, 37–42 CrossRef CAS.
- A. Erdohelyi, J. Cserenyi and F. Solymosi, J. Catal., 1993, 141, 287–299 CrossRef CAS.
- P. Ferreira-Aparicio, M. Fernandez-Garcia, A. Guerrero-Ruiz and I. Rodriguez-Ramos, J. Catal., 2000, 190, 296–308 CrossRef CAS.
- M. C. J. Bradford and M. A. Vannice, Catal. Rev., 1999, 41, 1–42 CAS.
- J. H. Bitter, K. Seshan and J. A. Lercher, Top. Catal., 2000, 10, 295–305 CrossRef CAS.
- S. M. Stagg, E. Romeo, C. Padro and D. E. Resasco, J. Catal., 1998, 178, 137–145 CrossRef CAS.
- M. Garcia-Dieguez, I. S. Pieta, M. C. Herrera, M. A. Larrubia, I. Malpartida and L. J. Alemany, Catal. Today, 2010, 149, 380–387 CrossRef CAS.
- Y. Ono, Appl. Catal., A, 1997, 155, 133–166 CrossRef CAS.
- D. C. Stoian, E. Taboada, J. Llorca, E. Molins, F. Medina and A. M. Segarra, Chem. Commun., 2013, 49, 5489–5491 RSC.
- D. Delledonne, F. Rivetti and U. Romano, Appl. Catal., A, 2001, 221, 241–251 CrossRef CAS.
- K. Tomishige, Y. Ikeda, T. Sakaihori and K. Fujimoto, J. Catal., 2000, 192, 355–362 CrossRef CAS.
- K. S. Lackner, Science, 2003, 300, 1677–1678 CrossRef CAS PubMed.
- G. Xiao, R. Singh, A. Chaffee and P. Webley, Int. J. Greenhouse Gas Control, 2011, 5, 634–639 CrossRef CAS.
- E. J. Swanson, K. J. Fricker, M. Sun and A.-H. A. Park, Phys. Chem. Chem. Phys., 2014, 16, 23440–23450 RSC.
- D. P. Butt, K. S. Lackner, C. H. Wendt, S. D. Conzone, H. Kung, Y.-C. Lu and J. K. Bremser, J. Am. Ceram. Soc., 1996, 79, 1892–1898 CrossRef CAS.
- S. Wang, S. Yan, X. Ma and J. Gong, Energy Environ. Sci., 2011, 4, 3805–3819 CAS.
- A. A. Olajire, J. Pet. Sci. Eng., 2013, 109, 364–392 CrossRef CAS.
- A. Auroux and A. Gervasini, J. Phys. Chem., 1990, 94, 6371–6379 CrossRef CAS.
- C. Pighini, T. Belin, J. Mijoin, P. Magnoux, G. Costentin and H. Lauron-Pernot, Appl. Surf. Sci., 2011, 257, 6952–6962 CrossRef CAS.
- J. Wang, L. Huang, R. Yang, Z. Zhang, J. Wu, Y. Gao, Q. Wang, D. O'Hare and Z. Zhong, Energy Environ. Sci., 2014, 7, 3478–3518 CAS.
- M. Zhao, A. I. Minett and A. T. Harris, Energy Environ. Sci., 2013, 6, 25–40 CAS.
- K. Vatopoulos and E. Tzimas, J. Cleaner Prod., 2012, 32, 251–261 CrossRef CAS.
- Y. Liang, D. P. Harrison, R. P. Gupta, D. A. Green and W. J. McMichael, Energy Fuels, 2004, 18, 569–575 CrossRef CAS.
- S. C. Lee, B. Y. Choi, T. J. Lee, C. K. Ryu, Y. S. Ahn and J. C. Kim, Catal. Today, 2006, 111, 385–390 CrossRef CAS.
- P. Gélin and M. Primet, Appl. Catal., B, 2002, 39, 1–37 CrossRef.
- V. C. Menon and S. Komarneni, J. Porous Mater., 1998, 5, 43–58 CrossRef CAS.
- S. Ma, D. Sun, J. M. Simmons, C. D. Collier, D. Yuan and H.-C. Zhou, J. Am. Chem. Soc., 2007, 130, 1012–1016 CrossRef PubMed.
- T. Nakayama, N. Ichikuni, S. Sato and F. Nozaki, Appl. Catal., A, 1997, 158, 185–199 CrossRef CAS.
- L.-C. Lin, A. H. Berger, R. L. Martin, J. Kim, J. A. Swisher, K. Jariwala, C. H. Rycroft, A. S. Bhown, M. Deem, M. Haranczyk and B. Smit, Nat. Mater., 2012, 11, 633–641 CrossRef CAS PubMed.
- M. Tagliabue, D. Farrusseng, S. Valencia, S. Aguado, U. Ravon, C. Rizzo, A. Corma and C. Mirodatos, Chem. Eng. J., 2009, 155, 553–566 CrossRef CAS.
- H. Furukawa, N. Ko, Y. B. Go, N. Aratani, S. B. Choi, E. Choi, A. Ö. Yazaydin, R. Q. Snurr, M. O'Keeffe, J. Kim and O. M. Yaghi, Science, 2010, 329, 424–428 CrossRef CAS PubMed.
- C. A. Grande and A. E. Rodrigues, Int. J. Greenhouse Gas Control, 2008, 2, 194–202 CAS.
- T.-H. Bae, M. R. Hudson, J. A. Mason, W. L. Queen, J. J. Dutton, K. Sumida, K. J. Micklash, S. S. Kaye, C. M. Brown and J. R. Long, Energy Environ. Sci., 2013, 6, 128–138 CAS.
- S. Xiang, Y. He, Z. Zhang, H. Wu, W. Zhou, R. Krishna and B. Chen, Nat. Commun., 2012, 3, 954 CrossRef PubMed.
- R. Krishna and J. M. van Baten, J. Membr. Sci., 2010, 360, 323–333 CrossRef CAS.
- M. P. Allen and D. J. Tildesley, Computer simulation of liquids, Clarendon Press, 1989 Search PubMed.
- A. Boutin, S. Couck, F.-X. Coudert, P. Serra-Crespo, J. Gascon, F. Kapteijn, A. H. Fuchs and J. F. M. Denayer, Microporous Mesoporous Mater., 2011, 140, 108–113 CrossRef CAS.
- F.-X. Coudert, C. Mellot-Draznieks, A. H. Fuchs and A. Boutin, J. Am. Chem. Soc., 2009, 131, 11329–11331 CrossRef CAS PubMed.
- P. A. Frey and A. D. Hegeman, Enzymatic Reaction Mechanisms, Oxford University Press, New York, 2007 Search PubMed.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef CAS PubMed.
- S. M. Whitney, R. L. Houtz and H. Alonso, Plant Physiol., 2011, 155, 27–35 CrossRef CAS PubMed.
- A. R. Portis and M. A. J. Parry, Photosynth. Res., 2007, 94, 121–143 CrossRef CAS PubMed.
- B. Kannappan and J. E. Gready, J. Am. Chem. Soc., 2008, 130, 15063–15080 CrossRef PubMed.
- C. C. Mann, Science, 1999, 283, 314–316 CrossRef CAS PubMed.
- I. Andersson, J. Mol. Biol., 1996, 259, 160–174 CrossRef CAS PubMed.
- R. A. Berner, Science, 1997, 276, 544–546 CrossRef CAS.
- P. N. Pearson and M. R. Palmer, Nature, 2000, 406, 695–699 CrossRef CAS PubMed.
- R. F. Sage, T. L. Sage and F. Kocacinar, Annu. Rev. Plant Biol., 2012, 63, 19–47 CrossRef CAS PubMed.
- R. Chollet, J. Vidal and M. H. O'Leary, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1996, 47, 273–298 CrossRef CAS PubMed.
- C. Peterhansel and V. G. Maurino, Plant Physiol., 2011, 155, 49–55 CrossRef CAS PubMed.
- B. A. Kimball, C. F. Morris, P. J. Pinter, G. W. Wall, D. J. Hunsaker, F. J. Adamsen, R. L. LaMorte, S. W. Leavitt, T. L. Thompson, A. D. Matthias and T. J. Brooks, New Phytol., 2001, 150, 295–303 CrossRef CAS.
- F.-W. Badeck, F. Rizza, C. Maré, L. Cattivelli, A. Zaldei and F. Miglietta, XVI convegno nazionale di Agrometeorologia, Firenze, 2013 Search PubMed.
- D. W. Lawlor and R. A. C. Mitchell, Plant, Cell Environ., 1991, 14, 807–818 CrossRef.
- P. M. Drennan and P. S. Nobel, Plant, Cell Environ., 2000, 23, 767–781 CrossRef CAS.
- A. G. Eric and S. N. Park, J. Exp. Bot., 1996, 47, 61–69 CrossRef.
- M. Calvin, Angew. Chem., Int. Ed., 1962, 1, 65–75 CrossRef.
- Jmol: an open-source Java viewer for chemical structures in 3D, http://www.jmol.org.
- B. Stec, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 18785–18790 CrossRef CAS PubMed.
- M. M. El-Hendawy, N. J. English and D. A. Mooney, J. Mol. Model., 2013, 19, 2329–2334 CrossRef CAS PubMed.
- Y. Kai, H. Matsumura and K. Izui, Arch. Biochem. Biophys., 2003, 414, 170–179 CrossRef CAS PubMed.
- I. P. Ting, Annu. Rev. Plant Physiol., 1985, 36, 595–622 CrossRef CAS.
- A. S. Raghavendra and R. F. Sage, C4 Photosynthesis and Related CO2 Concentrating Mechanisms, Springer Science & Business Media, 2010 Search PubMed.
- R. F. Sage, New Phytol., 2004, 161, 341–370 CrossRef CAS.
- H. Matsumura, Y. Xie, S. Shirakata, T. Inoue, T. Yoshinaga, Y. Ueno, K. Izui and Y. Kai, Structure, 2002, 10, 1721–1730 CrossRef CAS PubMed.
- J. K. Paulus, D. Schlieper and G. Groth, Nat. Commun., 2013, 4, 1518 CrossRef PubMed.
- I. P. Ting and C. B. Osmond, Plant Physiol., 1973, 51, 448–453 CrossRef CAS PubMed.
- Y. Kai, H. Matsumura, T. Inoue, K. Terada, Y. Nagara, T. Yoshinaga, A. Kihara, K. Tsumura and K. Izui, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 823–828 CrossRef CAS.
- O. E. Bläsing, P. Westhoff and P. Svensson, J. Biol. Chem., 2000, 275, 27917–27923 Search PubMed.
- C. P. Osborne and D. J. Beerling, Philos. Trans. R. Soc., B, 2006, 361, 173–194 CrossRef PubMed.
- V. Grignard, C. R. Acad. Sci., 1900, 130, 1322–1325 CAS.
- M. Orchin, J. Chem. Educ., 1989, 66, 586 CrossRef CAS.
- A. V. Nemukhin, I. A. Topol and F. Weinhold, Inorg. Chem., 1995, 34, 2980–2983 CrossRef CAS.
- R. W. Hoffmann and B. Holzer, Chem. Commun., 2001, 491–492 RSC.
- F. Blasberg, M. Bolte, M. Wagner and H.-W. Lerner, Organometallics, 2012, 31, 1001–1005 CrossRef CAS.
- A. M. Fracaroli, H. Furukawa, M. Suzuki, M. Dodd, S. Okajima, F. Gándara, J. A. Reimer and O. M. Yaghi, J. Am. Chem. Soc., 2014, 136, 8863–8866 CrossRef CAS PubMed.
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- V. Guillerm, F. Ragon, M. Dan-Hardi, T. Devic, M. Vishnuvarthan, B. Campo, A. Vimont, G. Clet, Q. Yang, G. Maurin, G. Férey, A. Vittadini, S. Gross and C. Serre, Angew. Chem., 2012, 51, 9267–9271 CrossRef CAS PubMed.
- Y. Fu, D. Sun, Y. Chen, R. Huang, Z. Ding, X. Fu and Z. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- D. Saha, T. Maity, S. Das and S. Koner, Dalton Trans., 2013, 42, 13912–13922 RSC.
- H. X. Deng, S. Grunder, K. E. Cordova, C. Valente, H. Furukawa, M. Hmadeh, F. Gandara, A. C. Whalley, Z. Liu, S. Asahina, H. Kazumori, M. O'Keeffe, O. Terasaki, J. F. Stoddart and O. M. Yaghi, Science, 2012, 336, 1018–1023 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- O. K. Farha, I. Eryazici, N. C. Jeong, B. G. Hauser, C. E. Wilmer, A. A. Sarjeant, R. Q. Snurr, S. T. Nguyen, A. Ö. Yazaydın and J. T. Hupp, J. Am. Chem. Soc., 2012, 134, 15016–15021 CrossRef CAS PubMed.
- H. Furukawa, Y. B. Go, N. Ko, Y. K. Park, F. J. Uribe-Romo, J. Kim, M. O'Keeffe and O. M. Yaghi, Inorg. Chem., 2011, 50, 9147–9152 CrossRef CAS PubMed.
- A. C. Kizzie, A. G. Wong-Foy and A. J. Matzger, Langmuir, 2011, 27, 6368–6373 CrossRef CAS PubMed.
- J. Liu, P. K. Thallapally, B. P. McGrail, D. R. Brown and J. Liu, Chem. Soc. Rev., 2012, 41, 2308–2322 RSC.
- Y. K. Hwang, D. Y. Hong, J. S. Chang, S. H. Jhung, Y. K. Seo, J. Kim, A. Vimont, M. Daturi, C. Serre and G. Férey, Angew. Chem., Int. Ed., 2008, 47, 4144–4148 CrossRef CAS PubMed.
- C. E. Wilmer, M. Leaf, C. Y. Lee, O. K. Farha, B. G. Hauser, J. T. Hupp and R. Q. Snurr, Nat. Chem., 2012, 4, 83–89 CrossRef CAS PubMed.
- A. L. Dzubak, L.-C. Lin, J. Kim, J. A. Swisher, R. Poloni, S. N. Maximoff, B. Smit and L. Gagliardi, Nat. Chem., 2012, 4, 810–816 CrossRef CAS PubMed.
- X. Q. Kong, H. X. Deng, F. Y. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2014, 341, 882–885 CrossRef PubMed.
- D. Banerjee and J. B. Parise, Cryst. Growth Des., 2011, 11, 4704–4720 CAS and references therein.
- M. Dinča and J. R. Long, J. Am. Chem. Soc., 2005, 127, 9376–9377 CrossRef PubMed.
- L. Han, L. Qin, X. Z. Yan, L. P. Xu, J. L. Sun, L. Yu, H. B. Chen and X. D. Zou, Cryst. Growth Des., 2013, 13, 1807–1811 CAS.
- Y. Liu, Y.-P. Chen, T.-F. Liu, A. A. Yakovenko, A. M. Raiff and H.-C. Zhou, CrystEngComm, 2013, 15, 9688–9693 RSC.
- D. Britt, H. Furukawa, B. Wang, T. G. Glover and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 20637–20640 CrossRef CAS PubMed.
- J. G. Vitillo, L. Regli, S. Chavan, G. Ricchiardi, G. Spoto, P. D. C. Dietzel, S. Bordiga and A. Zecchina, J. Am. Chem. Soc., 2008, 130, 8386–8396 CrossRef CAS PubMed.
- C. Prestipino, L. Regli, J. G. Vitillo, F. Bonino, A. Damin, C. Lamberti, A. Zecchina, P. L. Solari, K. O. Kongshaug and S. Bordiga, Chem. Mater., 2006, 18, 1337–1346 CrossRef CAS.
- P. D. C. Dietzel, V. Besikiotis and R. Blom, J. Mater. Chem., 2009, 19, 7362–7370 RSC.
- W. L. Queen, C. M. Brown, D. K. Britt, P. Zajdel, M. R. Hudson and O. M. Yaghi, J. Phys. Chem. C, 2011, 115, 24915–24919 CAS.
- W. Zhou, H. Wu and T. Yildirim, J. Am. Chem. Soc., 2008, 130, 15268–15269 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, G. Turnes Palomino, C. Otero Areán and S. Bordiga, J. Phys. Chem. C, 2010, 114, 11185–11191 CAS.
- A. Pulido, M. R. Delgado, O. Bludsky, M. Rubes, P. Nachtigall and C. O. Arean, Energy Environ. Sci., 2009, 2, 1187–1195 CAS.
- Y. F. Chen, A. Nalaparaju, M. Eddaoudi and J. W. Jiang, Langmuir, 2012, 28, 3903–3910 CrossRef CAS PubMed.
- T. M. McDonald, W. R. Lee, J. A. Mason, B. M. Wiers, C. S. Hong and J. R. Long, J. Am. Chem. Soc., 2012, 134, 7056–7065 CrossRef CAS PubMed.
- S. Choi, T. Watanabe, T.-H. Bae, D. S. Sholl and C. W. Jones, J. Phys. Chem. Lett., 2012, 3, 1136–1141 CrossRef CAS PubMed.
- J. Yu and P. B. Balbuena, J. Phys. Chem. C, 2013, 117, 3383–3388 CAS.
- Y. E. Cheon, J. Park and M. P. Suh, Chem. Commun., 2009, 5436–5438 RSC.
- Y. Cao, F. Song, Y. Zhao and Q. Zhong, J. Environ. Sci., 2013, 25, 2081–2087 CrossRef CAS.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocella, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef CAS PubMed.
- D.-Y. Hong, Y. K. Hwang, C. Serre, G. Férey and J.-S. Chang, Adv. Funct. Mater., 2009, 19, 1537–1552 CrossRef CAS.
- A. Demessence, D. M. D'Alessandro, M. L. Foo and J. R. Long, J. Am. Chem. Soc., 2009, 131, 8784–8785 CrossRef CAS PubMed.
- J. C. Hicks, J. H. Drese, D. J. Fauth, M. L. Gray, G. G. Qi and C. W. Jones, J. Am. Chem. Soc., 2008, 130, 2902–2903 CrossRef CAS PubMed.
- J. Ethiraj, E. Albanese, B. Civalleri, J. G. Vitillo, F. Bonino, S. Chavan, G. C. Shearer, K. P. Lillerud and S. Bordiga, ChemSusChem, 2014, 7, 3382–3388 CrossRef CAS PubMed.
- K. L. Gurunatha, K. Uemura and T. K. Maji, Inorg. Chem., 2008, 47, 6578–6580 CrossRef CAS PubMed.
- A. Rossin, A. Ienco, F. Costantino, T. Montini, B. Di Credico, M. Caporali, L. Gonsalvi, P. Fornasiero and M. Peruzzini, Cryst. Growth Des., 2008, 8, 3302–3308 CAS.
- J. A. Rood, B. C. Noll and K. W. Henderson, Inorg. Chem., 2006, 45, 5521–5528 CrossRef CAS PubMed.
- A. Mallick, S. Saha, P. Pachfule, S. Roy and R. Banerjee, Inorg. Chem., 2011, 50, 1392–1401 CrossRef CAS PubMed.
- A. Rossin, M. R. Chierotti, G. Giambastiani, R. Gobetto and M. Peruzzini, CrystEngComm, 2012, 14, 4454–4460 RSC.
- A. Mallick, S. Saha, P. Pachfule, S. Roy and R. Banerjee, J. Mater. Chem., 2010, 20, 9073–9080 RSC.
- P. D. C. Dietzel, R. Blom and H. Fjellvåg, Eur. J. Inorg. Chem., 2008, 2008, 3624–3632 CrossRef.
- Z. R. Herm, J. A. Swisher, B. Smit, R. Krishna and J. R. Long, J. Am. Chem. Soc., 2011, 133, 5664–5667 CrossRef CAS PubMed.
- J. M. C. Plane and C. L. Whalley, J. Phys. Chem. A, 2012, 116, 6240–6252 CrossRef CAS PubMed.
- J. J. Olivero and G. E. Thomas, Adv. Space Res., 2001, 28, 931–936 CrossRef CAS.
- J. M. C. Plane, Chem. Rev., 2003, 103, 4963–4984 CrossRef CAS PubMed.
- N. McBride, S. F. Green and J. A. M. McDonnell, Adv. Space Res., 1999, 23, 73–82 CrossRef.
- B. Mason, Handbook of elemental abundances of the elements in meteorites, Gordon and Breach, Newark, NJ, 1971 Search PubMed.
- W. Swider, Planet. Space Sci., 1984, 32, 307–312 CrossRef CAS.
- M. Scharringhausen, Investigation of Mesospheric and Thermospheric Magnesium Species from Space, VDM Verlag Dr. Mueller e. K, Saarbrücken, Germany, 2007 Search PubMed.
- J. M. C. Plane, C. S. Gardner, J. Yu, C. Y. She, R. R. Garcia and H. C. Pumphrey, J. Geophys. Res., 1999, 104, 3773–3788 CrossRef CAS.
- S. Campbell, P. Hollins, E. McCash and M. W. Roberts, J. Electron Spectrosc. Relat. Phenom., 1986, 39, 145–153 CrossRef CAS.
- R. G. Copperthwaite, P. R. Davies, M. A. Morris, M. W. Roberts and R. A. Ryder, Catal. Lett., 1988, 1, 11–19 CrossRef CAS.
- M. B. Jensen, L. G. M. Pettersson, O. Swang and U. Olsbye, J. Phys. Chem. B, 2005, 109, 16774–16781 CrossRef CAS PubMed.
- H. Kawakami and S. Yoshida, J. Chem. Soc., Faraday Trans., 1984, 80, 921–932 RSC.
- L. Hopkinson, P. Kristova, K. Rutt and G. Cressey, Geochim. Cosmochim. Acta, 2012, 76, 1–13 CrossRef CAS.
- B.-J. Kim, K.-S. Cho and S.-J. Park, J. Colloid Interface Sci., 2010, 342, 575–578 CrossRef CAS PubMed.
- W. H. Cheng and H. H. Kung, Surf. Sci., 1981, 102, L21–L28 CrossRef CAS.
- E. N. Gribov, S. Bertarione, D. Scarano, C. Lamberti, G. Spoto and A. Zecchina, J. Phys. Chem. B, 2004, 108, 16174–16186 CrossRef CAS.
- G. Spoto, E. N. Gribov, G. Ricchiardi, A. Damin, D. Scarano, S. Bordiga, C. Lamberti and A. Zecchina, Progr. Surf. Sci., 2004, 76, 71–146 CrossRef CAS.
- M.-L. Bailly, G. N. Costentin, H. L. N. Lauron-Pernot, J. M. Krafft and M. Che, J. Phys. Chem. B, 2004, 109, 2404–2413 CrossRef PubMed.
- C. Chizallet, G. Costentin, M. Che, F. Delbecq and P. Sautet, J. Phys. Chem. B, 2006, 110, 15878–15886 CrossRef CAS PubMed.
- E. Knözinger, K.-H. Jacob, S. Singh and P. Hofmann, Surf. Sci., 1993, 290, 388–402 CrossRef.
- C. Chizallet, G. Costentin, M. Che, F. Delbecq and P. Sautet, J. Am. Chem. Soc., 2007, 129, 6442–6452 CrossRef CAS PubMed.
- Y. Fukuda and K. Tanabe, Bull. Chem. Soc. Jpn., 1973, 46, 1616–1619 CrossRef CAS.
- R. A. Robie, B. S. Hemingway and J. R. Fisher, Thermodynamic properties of minerals and related substances at 298.15 K and 1 bar (105 pascals) pressure and at high temperatures, Bulletin 1452, U.S. Geological Survey, Washington DC, 1984 Search PubMed.
- K. Nakagawa and T. Ohashi, J. Electrochem. Soc., 1998, 145, 1344–1346 CrossRef CAS.
- R. V. Siriwardane, Regenerable Sorbents for CO2 Capture from Moderate and High Temperature Applications, U.S. Pat., 7,314,847, 2008.
- J. Roggenbuck, G. Koch and M. Tiemann, Chem. Mater., 2006, 18, 4151–4156 CrossRef CAS.
- J. Roggenbuck and M. Tiemann, J. Am. Chem. Soc., 2005, 127, 1096–1097 CrossRef CAS PubMed.
- K. K. Han, Y. Zhou, Y. Chun and J. H. Zhu, J. Hazard. Mater., 2012, 203, 341–347 CrossRef PubMed.
- Z. Zhao, H. Dai, Y. Du, J. Deng, L. Zhang and F. Shi, Mater. Chem. Phys., 2011, 128, 348–356 CrossRef CAS.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175–181 CrossRef CAS.
- R. Das, P. Pachfule, R. Banerjee and P. Poddar, Nanoscale, 2012, 4, 591–599 RSC.
- S.-W. Bian, J. Baltrusaitis, P. Galhotra and V. H. Grassian, J. Mater. Chem., 2010, 20, 8705–8710 RSC.
- M. Bhagiyalakshmi, J. Y. Lee and H. T. Jang, Int. J. Greenhouse Gas Control, 2010, 4, 51–56 CrossRef CAS.
- K. K. Han, Y. Zhou, W. G. Lin and J. H. Zhu, Microporous Mesoporous Mater., 2013, 169, 112–119 CrossRef CAS.
- H. N. Li, L. Zhang, H. X. Dai and H. He, Inorg. Chem., 2009, 48, 4421–4434 CrossRef CAS PubMed.
- W.-J. Liu, H. Jiang, K. Tian, Y.-W. Ding and H.-Q. Yu, Environ. Sci. Technol., 2013, 47, 9397–9403 CrossRef CAS PubMed.
- Y. Wang, J. H. Zhu, J. M. Cao, Y. Chun and Q. H. Xu, Microporous Mesoporous Mater., 1998, 26, 175–184 CrossRef CAS.
- J. H. Zhu, Y. Wang, Y. Chun, Z. Xing and Q. H. Xu, Mater. Lett., 1998, 35, 177–182 CrossRef CAS.
- D. Barthomeuf, Catal. Rev., 1996, 38, 521–612 Search PubMed.
- Y. L. Wei, Y. M. Wang, J. H. Zhu and Z. Y. Wu, Adv. Mater., 2003, 15, 1943–1945 CrossRef CAS.
- J. C. Mabry and K. Mondal, Environ. Technol., 2011, 32, 55–67 CrossRef CAS PubMed.
- X. C. Yan, H. Guo, D. J. Yang, S. L. Qiu and X. D. Yao, Curr. Org. Chem., 2014, 18, 1335–1345 CrossRef CAS.
- Z. Kowalczyk, K. Stolecki, W. Rarog-Pilecka, E. B. Miskiewicz, E. Wilczkowska and Z. Karpiniski, Appl. Catal., A, 2008, 342, 35–39 CrossRef CAS.
- H. Y. Kim, H. M. Lee and J.-N. Park, J. Phys. Chem. C, 2010, 114, 7128–7131 CAS.
- S. Mori, W. C. Xu, T. Ishidzuki, N. Ogasawara, J. Imai and K. Kobayashi, Appl. Catal., A, 1996, 137, 255–268 CrossRef CAS.
- J. Rostrup-Nielsen, U.S. Pat., 3,791,993, 1974.
- J. Zhang, H. Wang, C. Xi, M. Shakouri, Y. Hu and A. K. Dalai, in Nanocatalysis for Fuels and Chemicals, American Chemical Society, 2012, vol. 1092, pp. 195–221 Search PubMed.
- H. Y. Wang and E. Ruckenstein, Appl. Catal., A, 2000, 204, 143–152 CrossRef CAS.
- K. Nagaoka, M. Okamura and K.-I. Aika, Catal. Commun., 2001, 2, 255–260 CrossRef CAS.
- A. Ballarini, F. Basile, P. Benito, I. Bersani, G. Fornasari, S. de Miguel, S. C. P. Maina, J. Vilella, A. Vaccari and O. A. Scelza, Appl. Catal., A, 2012, 433, 1–11 CrossRef.
- L. Desgranges, G. Calvarin and G. Chevrier, Acta Crystallogr., 1996, 52, 82–86 Search PubMed.
- M. Bellotto, B. Rebours, O. Clause, J. Lynch, D. Bazin and E. Elkaïm, J. Phys. Chem., 1996, 100, 8527–8534 CrossRef CAS.
- S.-H. Hong, M.-S. Jang, S. J. Cho and W.-S. Ahn, Chem. Commun., 2014, 50, 4927–4930 RSC.
- G. R. Williams and D. O'Hare, J. Mater. Chem., 2006, 16, 3065–3074 RSC.
- V. Rives and S. Kannan, J. Mater. Chem., 2000, 10, 489–495 RSC.
- Q. Wang, H. H. Tay, Z. Zhong, J. Luo and A. Borgna, Energy Environ. Sci., 2012, 5, 7526–7530 CAS.
- G. Fetter, M. T. OlguÃn, P. Bosch and S. Bulbulian, J. Porous Mater., 2000, 7, 469–473 CrossRef CAS.
- A. Garcia-Gallastegui, D. Iruretagoyena, V. Gouvea, M. Mokhtar, A. M. Asiri, S. N. Basahel, S. A. Al-Thabaiti, A. O. Alyoubi, D. Chadwick and M. S. P. Shaffer, Chem. Mater., 2012, 24, 4531–4539 CrossRef CAS.
- S. Choi, J. H. Drese and C. W. Jones, ChemSusChem, 2009, 2, 796–854 CrossRef CAS PubMed.
- E. Ochoa-Fernandez, H. K. Rusten, H. A. Jakobsen, M. Ronning, A. Holmen and D. Chen, Catal. Today, 2005, 106, 41–46 CrossRef CAS.
- C. T. Yavuz, B. D. Shinall, A. V. Iretskii, M. G. White, T. Golden, M. Atilhan, P. C. Ford and G. D. Stucky, Chem. Mater., 2009, 21, 3473–3475 CrossRef CAS.
- A. F. Lucredio, J. M. Assaf and E. M. Assaf, Appl. Catal., A, 2011, 400, 156–165 CrossRef CAS.
- Y. Gao, Z. Zhang, J. Wu, X. Yi, A. Zheng, A. Umar, D. O'Hare and Q. Wang, J. Mater. Chem. A, 2013, 1, 12782–12790 CAS.
- D. Aymes, D. Ballivet-Tkatchenko, K. Jeyalakshmi, L. Saviot and S. Vasireddy, Catal. Today, 2009, 147, 62–67 CrossRef CAS.
- A. Sanna, M. Uibu, G. Caramanna, R. Kuusik and M. M. Maroto-Valer, Chem. Soc. Rev., 2014, 43, 8049–8080 RSC.
- H. Gies and H. van Koningsveld, Catalog of Disorder in Zeolite Frameworks, http://www.iza-structure.org/databases/ Search PubMed.
- P. Nachtigall, L. Grajciar, J. Perez-Pariente, A. B. Pinar, A. Zukal and J. Cejka, Phys. Chem. Chem. Phys., 2012, 14, 1117–1120 RSC.
- F. Su and C. Lu, Energy Environ. Sci., 2012, 5, 9021–9027 CAS.
- Q. Liu, T. Pham, M. D. Porosoff and R. F. Lobo, ChemSusChem, 2012, 5, 2237–2242 CrossRef CAS PubMed.
- J. Zhang, R. Singh and P. A. Webley, Microporous Mesoporous Mater., 2008, 111, 478–487 CrossRef CAS.
- M. Palomino, A. Corma, F. Rey and S. Valencia, Langmuir, 2009, 26, 1910–1917 CrossRef PubMed.
- T. Remy, S. A. Peter, L. Van Tendeloo, S. Van der Perre, Y. Lorgouilloux, C. E. A. Kirschhock, G. V. Baron and J. F. M. Denayer, Langmuir, 2013, 29, 4998–5012 CrossRef CAS PubMed.
- S. Y. Zhang, O. Talu and D. T. Hayhurst, J. Phys. Chem., 1991, 95, 1722–1726 CrossRef CAS.
- J. J. Pluth and J. V. Smith, J. Am. Chem. Soc., 1980, 102, 4704–4708 CrossRef CAS.
- J. Liu, Y. Wang, A. I. Benin, P. Jakubczak, R. R. Willis and M. D. LeVan, Langmuir, 2010, 26, 14301–14307 CrossRef CAS PubMed.
- O. Zavorotynska, J. G. Vitillo, G. Spoto and A. Zecchina, Int. J. Hydrogen Energy, 2011, 36, 7944–7950 CrossRef CAS.
- C. Pazé, S. Bordiga, C. Lamberti, M. Salvalaggio, A. Zecchina and G. Bellussi, J. Phys. Chem. B, 1997, 101, 4740–4751 CrossRef.
- Y.-K. Kim, Y.-H. Mo, J. Lee, H.-S. You, C.-K. Yi, Y. C. Park and S.-E. Park, J. Nanosci. Nanotechnol., 2013, 13, 2703–2707 CrossRef CAS PubMed.
- H. Tsuji, F. Yagi, H. Hattori and H. Kita, in Studies in Surface Science and Catalysis, ed. L. Guczi, F. Solymosi and P. Tétényi, Elsevier, Budapest, 1993, vol. 75, pp. 1171–1183 Search PubMed.
- J. G. Vitillo, et al., J. Mater. Chem., 2015 Search PubMed , in preparation.
- F. Bergaya, B. K. G. Theng and G. Lagaly, Developments in Clay Science in Handbook of clay science, Elsevier, Oxford, 2006 Search PubMed.
- C. Chen, D.-W. Park and W.-S. Ahn, Appl. Surf. Sci., 2013, 283, 699–704 CrossRef CAS.
- F. Bardelli, C. Mondelli, M. Didier, J. G. Vitillo, D. R. Cavicchia, J.-C. Robinet, L. Leone and L. Charlet, Appl. Geochem., 2014, 49, 168–177 CrossRef CAS.
- C. Mondelli, F. Bardelli, J. G. Vitillo, M. Didier, J. Brendle, D. R. Cavicchia, J.-C. Robinet and L. Charlet, Int. J. Hydrogen Energy, 2015, 40, 2698–2709 CrossRef CAS.
- W. Wang, X. Wang, C. Song, X. Wei, J. Ding and J. Xiao, Energy Fuels, 2013, 27, 1538–1546 CrossRef CAS.
- P. B. Kelemen and J. R. Matter, Proc. Natl. Acad. Sci. U. S. A., 2008, 17295–17300 CrossRef CAS.
- W. K. O'Connor, D. C. Dahlin, G. E. Rush, C. L. Dahlin and W. K. Collins, Miner. Metall. Process., 2002, 19, 95–101 Search PubMed.
- S. J. Gerdemann, W. K. O'Connor, D. C. Dahlin, L. R. Penner and H. Rush, Environ. Sci. Technol., 2007, 41, 2587–2593 CrossRef CAS PubMed.
- M. J. McKelvy, A. V. G. Chizmeshya, J. Diefenbacher, H. Béarat and G. Wolf, Environ. Sci. Technol., 2004, 38, 6897–6903 CrossRef CAS PubMed.
- F. Larachi, I. Daldoul and G. Beaudoin, Geochim. Cosmochim. Acta, 2010, 74, 3051–3075 CrossRef CAS.
- H. C. Oskierski, B. Z. Dlugogorski and G. Jacobsen, Chem. Geol., 2013, 358, 156–169 CrossRef CAS.
- F. Larachi, J. P. Gravel, B. P. A. Grandjean and G. Beaudoin, Int. J. Greenhouse Gas Control, 2012, 6, 69–76 CrossRef CAS.
- S. Chatterjee, S. Sengupta, T. Saha-Dasgupta, K. Chatterjee and N. Mandal, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115103 CrossRef.
- D. Gournis, A. Lappas, M. A. Karakassides, D. Többens and A. Moukarika, Phys. Chem. Miner., 2008, 35, 49–58 CrossRef CAS.
- B. Fubini and C. Otero Arean, Chem. Soc. Rev., 1999, 28, 373–381 RSC.
- F. Turci, M. Tomatis, S. Mantegna, G. Cravotto and B. Fubini, J. Environ. Monit., 2007, 9, 1064–1066 RSC.
- R. Lafay, G. Montes-Hernandez, E. Janots, R. Chiriac, N. Findling and F. Toche, Chem.–Eur. J., 2013, 19, 5417–5424 CrossRef CAS PubMed.
- A. K. Chakraborty, K. B. Bischoff, G. Astarita and J. R. Damewood, J. Am. Chem. Soc., 1988, 110, 6947–6954 CrossRef CAS.
- N. P. Stadie, E. Callini, B. Richter, T. R. Jensen, A. Borgschulte and A. Züttel, J. Am. Chem. Soc., 2014, 136, 8181–8184 CrossRef CAS PubMed.
- B. T. Thompson, T. N. Gallaher and T. C. DeVore, Polyhedron, 1983, 2, 619–621 CrossRef CAS.
- C. L. Hugelshofer, A. Borgschulte, E. Callini, S. K. Matam, J. Gehrig, D. T. Hog and A. Züttel, J. Phys. Chem. C, 2014, 118, 15940–15945 CAS.
- H. Ando, M. Fujiwara, Y. Matsumura, H. Miyamura, H. Tanaka and Y. Souma, J. Alloys Compd., 1995, 223, 139–141 CrossRef CAS.
- E. Akiba, Y. Y. Ishido, H. Hayakawa, S. Shin and K. Nomura, Z. Phys. Chem., 1989, 164, 1319–1324 CrossRef.
- A. A. Peterson, F. Abild-Pedersen, F. Studt, J. Rossmeisl and J. K. Norskov, Energy Environ. Sci., 2010, 3, 1311–1315 CAS.
- K. J. Williams, A. B. Boffa, M. Salmeron, A. T. Bell and G. A. Somorjai, Catal. Lett., 1991, 9, 415–426 CrossRef CAS.
- J. Ye, C. Liu and Q. Ge, J. Phys. Chem. C, 2012, 116, 7817–7825 CAS.
- M. Tian, Master thesis, University of East Anglia, 2010.
- S. Bouaricha, J. Huot, D. Guay and R. Schulz, Int. J. Hydrogen Energy, 2002, 27, 909–913 CrossRef CAS.
- A. S. Pedersen and B. Larsen, Int. J. Hydrogen Energy, 1993, 18, 297–300 CrossRef CAS.
- P. M. Zimmerman, Z. Zhang and C. B. Musgrave, Inorg. Chem., 2010, 49, 8724–8728 CrossRef CAS PubMed.
- J. Zhang, Y. Zhao, D. L. Akins and J. W. Lee, J. Phys. Chem. C, 2011, 115, 8386–8392 CAS.
- R. Xiong, J. S. Zhang and J. W. Lee, J. Phys. Chem. C, 2013, 117, 3799–3803 CAS.
- J. S. Zhang and J. W. Lee, Carbon, 2013, 53, 216–221 CrossRef CAS.
- J. S. Zhang, A. Byeon and J. W. Lee, J. Mater. Chem. A, 2013, 1, 8665–8671 CAS.
- J. S. Zhang, Y. Zhao, X. D. Guan, R. E. Stark, D. L. Akins and J. W. Lee, J. Phys. Chem. C, 2012, 116, 2639–2644 CAS.
- T. Wartik and R. K. Pearson, J. Inorg. Nucl. Chem., 1958, 7, 404–411 CrossRef CAS.
- G. W. Gribble and C. F. Nutaitis, Org. Prep. Proced. Int., 1985, 17, 317–384 CrossRef CAS.
- J. G. Burr, W. G. Brown and H. E. Heller, J. Am. Chem. Soc., 1950, 72, 2560–2562 CrossRef CAS.
- R. Xiong, J. Zhang, Y. Zhao, D. L. Akins and J. W. Lee, Int. J. Hydrogen Energy, 2012, 37, 3344–3349 CrossRef CAS.
- Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov and H. Hagemann, Angew. Chem., Int. Ed., 2011, 50, 11162–11166 CrossRef CAS PubMed.
- A. Zűttel, Proceedings of the MH2014 Conference, Manchester, UK, 2014 Search PubMed.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS PubMed.
- J. Rabone, Y. F. Yue, S. Y. Chong, K. C. Stylianou, J. Bacsa, D. Bradshaw, G. R. Darling, N. G. Berry, Y. Z. Khimyak, A. Y. Ganin, P. Wiper, J. B. Claridge and M. J. Rosseinsky, Science, 2010, 329, 1053–1057 CrossRef CAS PubMed.
- A. Schneemann, V. Bon, I. Schwedler, I. Senkovska, S. Kaskel and R. A. Fischer, Chem. Soc. Rev., 2014, 43, 6062–6096 RSC.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373–1382 CrossRef CAS PubMed.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- C. Volkringer, M. Meddouri, T. Loiseau, N. Guillou, J. Marrot, G. Ferey, M. Haouas, F. Taulelle, N. Audebrand and M. Latroche, Inorg. Chem., 2008, 47, 11892–11901 CrossRef CAS PubMed.
- C. Petit, J. Burress and T. J. Bandosz, Carbon, 2011, 49, 563–572 CrossRef CAS.
- C. Petit and T. J. Bandosz, Adv. Funct. Mater., 2011, 21, 2108–2117 CrossRef CAS.
- Y. Zhao, M. Seredych, Q. Zhong and T. J. Bandosz, RSC Adv., 2013, 3, 9932–9941 RSC.
- L. Hou, W.-J. Shi, Y.-Y. Wang, Y. Guo, C. Jin and Q.-Z. Shi, Chem. Commun., 2011, 47, 5464–5466 RSC.
- C. Chen, S. T. Yang, W. S. Ahn and R. Ryoo, Chem. Commun., 2009, 3627–3629 RSC.
- G. K. Parshetti, S. Chowdhury and R. Balasubramanian, RSC Adv., 2014, 4, 44634–44643 RSC.
- J. Song, W. Shen, J. Wang and W. Fan, Carbon, 2014, 69, 255–263 CrossRef CAS.
- S. H. Lamlom and R. A. Savidge, Biomass Bioenergy, 2003, 25, 381–388 CrossRef CAS.
- http://www.extension.org/pages/70456/poplar-populus-spp-trees-for-biofuel-production#.VGfRr_mG98F, 2013.
- http://www.arsia.toscana.it/UserFiles/File/Woodland/Risultati_di_un_progetto_triennale_finanziato_dalla_CCIAA_di_Padova.pdf, 2014.
- A. Brunori, Legno ed energia: come produrre energie con le biomasse agricole (in italian), Il Sole 24 Ore Edagricole, 2008 Search PubMed.
- E. N. Speelman, M. M. L. Van Kempen, J. Barke, H. Brinkhuis, G. J. Reichart, A. J. P. Smolders, J. G. M. Roelofs, F. Sangiorgi, J. W. De Leeuw, A. F. Lotter and J. S. Sinninghe Damsté, Geobiology, 2009, 7, 155–170 CrossRef CAS PubMed.
- H. Brinkhuis, S. Schouten, M. E. Collinson, A. Sluijs, J. S. S. Damsté, G. R. Dickens, M. Huber, T. M. Cronin, J. Onodera, K. Takahashi, J. P. Bujak, R. Stein, J. van der Burgh, J. S. Eldrett, I. C. Harding, A. F. Lotter, F. Sangiorgi, H. V. K.-V. Cittert, J. W. de Leeuw, J. Matthiessen, J. Backman, K. Moran and S. the Expedition, Nature, 2006, 441, 606–609 CrossRef CAS PubMed.
- D.-J. Shi and D. Hall, Bot. Rev., 1988, 54, 353–386 CrossRef.
- M. S. A. Rahaman, L.-H. Cheng, X.-H. Xu, L. Zhang and H.-L. Chen, Renewable Sustainable Energy Rev., 2011, 15, 4002–4012 CrossRef.
- Y.-B. Seo, Y.-W. Lee, C.-H. Lee and H.-C. You, Bioresour. Technol., 2010, 101, 2549–2553 CrossRef CAS PubMed.
- http://theazollafoundation.org/, 2014.
- M. H. Yoon, Y. W. Lee, C. H. Lee and Y. B. Seo, Bioresour. Technol., 2012, 126, 198–201 CrossRef CAS PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- K. Ashworth, O. Wild and C. N. Hewitt, Nat. Clim. Change, 2013, 3, 492–496 CrossRef CAS.
- D. Taraborrelli, M. G. Lawrence, J. N. Crowley, T. J. Dillon, S. Gromov, C. B. M. Grosz, L. Vereecken and J. Lelieveld, Nat. Geosci., 2011, 5, 190–193 CrossRef.
- A. T. Archibald, Nat. Geosci., 2011, 4, 659–660 CrossRef CAS.
- S. von Caemmerer, W. P. Quick and R. T. Furbank, Science, 2012, 336, 1671–1672 CrossRef CAS PubMed.
- S. Rachmilevitch, A. B. Cousins and A. J. Bloom, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11506–11510 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the calculations for the quantities reported in Fig. 2 and in Table 8; comprehensive list of all the Mg-based metal organic frameworks reported in the literature. See DOI: 10.1039/c5ra02835c |
| This journal is © The Royal Society of Chemistry 2015 |