
Enhanced photocatalytic activities of Bi2WO6 by introducing Zn to replace Bi lattice sites: a first-principles study†
Abstract
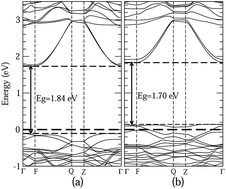
The geometry structure, electronic structure, and band edge positions of Zn-doped Bi2WO6 have been studied using a first-principles method. Bi1.75Zn0.25WO6 has a high quantum efficiency (QE) caused by the large mobility along different orientations and the efficient photogenerated-electron trap. It exhibits an enhanced visible-light absorption capability (α) due to the large increase of the density of electrons in the valence band maximum (VBM) and decrease in the band gap. The interactions are both strengthened between layers and among atoms within (Bi2O2)n layers. These strengthened interactions induce an enhanced stereochemically active Bi lone pair effect, which is identified as the cause of its unique electronic structure. Furthermore, the valence band (VB) and conduction band (CB) edges of Zn-doped Bi2WO6 are slightly shifted upwards. This indicates that the dominant active species during photocatalytic reaction for Bi1.75Zn0.25WO6 are not only hole and electron but also ˙O2− ion and ˙OH radical.
 Please wait while we load your content...
Please wait while we load your content...

