
Planar wheel-type M©BnHn2−/−/0 clusters (M = Cr, Mn and Fe for dianion, anion and neutral, respectively; n = 6 and 7)
Jianhua Hou*ab,
Qian Duanab,
Jieming Qinabc,
Xiande Shenab,
Jianxun Zhaoab,
Qingcheng Liangab,
Dayong Jiangab and
Shang Gaoab
aSchool of Materials Science and Engineering, Changchun University of Science and Technology, Changchun 130022, People’s Republic of China. E-mail: houjh163@163.com
bEngineering Research Center of Optoelectronic Functional Materials, Ministry of Education, People’s Republic of China
cInner Mongolia University For The Nationalities, People’s Republic of China
First published on 9th April 2015
Abstract
We adopted a new “electronic” strategy that is adding two electrons into the dz2 orbital of the central M atom to form a lone pair, which is in contrast to the Hoffmann’s “electronic” strategy that is delocalizing the lone pair on the center atom, to turn the bowl-type MBnHn0/+ (M = Cr and Mn; n = 6 and 7) clusters into planar wheel-type M©BnHn2−/− clusters. Their isoelectronic neutral clusters, Fe©B6H6 and Fe©B7H7, also possess planar wheel-type structures. The large binding energy, vertical ionization potential, and vertical electron affinity indicate that the planar wheel-type M©BnHn2−/− clusters are chemically stable. This study may open up a new area in coordination chemistry for planar hexa- and hepta-coordinate transition metal and we expect further experimental explorations of synthesis and potential applications.
1. Introduction
In the 1970s, Hoffmann et al.1 pointed out that the electronic structure of planar D4h CH4 is unfavorable due to a nonbonding pz-orbital lone pair on the planar tetracoordinate carbon (ptC). Based on this analysis, they suggested that the ptC arrangements could be achieved by using an “electronic” strategy, that is, delocalizing the lone pair by a π acceptor. Consequently, many ptC clusters, designed using “electronic” strategies, have been characterized theoretically and experimentally.2–4 Since then, planar tetra-, penta-, hexa-, hepta- and octacoordinate main group elements have been investigated extensively.5–11 However, these extreme violations of classical bonding principles are by no means limited to the main group elements. Numerous planar wheel-type clusters with a central hypercoordinate transition metal (M) encircled by different ligands, such as planar n-membered metal rings (n = 5 to 7)12–15 and planar n-membered boron rings (n = 7 to 10),16–22 have been studied experimentally and computationally. The studies from M©Bn (ref. 16 and 22) (the © sign, proposed by Wang and Boldyrev,18 designates a planar n-membered boron ring with a central M atom and is distinct from the @ sign used for a 3D cage cluster with an endohedral atom) demonstrate that transition metal atoms are more suitable for the central position in the boron ring, because they prefer to participate in delocalized bonding with the boron ring. To achieve the design of M©Bnk−, Wang and Boldyrev also developed a chemical bonding model in which to satisfy the peripheral B–B bonding and the delocalized σ and π bonding, the required valence for the central transition-metal atom M, is 12 − n − k.17–19Compared to pure boron clusters, hydrogenated small boron clusters favor adopting 3D aromatic structures.23–26 Thus, it is challenging to obtain planar hypercoordinate elements with BH ligands. In 2009, Yu and co-workers27 reported a planar wheel-type D5h B©B5H5+ cluster, which is the global minimum according to their calculation. In 2013, Cheng’s group designed some MBnHn0/+ (M = Cr and Mn; n = 6 and 7) clusters.28 Both Cr and Mn+ have six valence electrons which were expected to occupy the empty overlapped pz orbitals of B atoms to obtain the planar wheel type clusters in their assumption. However, the MBnHn0/+ clusters are bowl-type structures and do not reach the planar wheel-type structures due to participation of the dz2 orbital of the M atoms in the overlap between the dxz and dyz orbitals of the M atoms and the pz orbitals of the B atoms. Here, we adopt a new “electronic” strategy that is adding two electrons into the dz2 orbital to form a lone pair, which is in contrast to the “electronic” strategy of Hoffmann,1 to achieve planar wheel-type M©BnHn2−/− clusters. Density functional theory (DFT) was applied to investigate the geometrical and electronic structures of the planar wheel-type M©BnHn2−/− clusters as well as their isoelectronic neutral clusters, Fe©B6H6 and Fe©B7H7. The results show that the planar wheel-type M©BnHn2−/−/0 (M = Cr, Mn and Fe for dianion, anion and neutral respectively; n = 6 and 7) clusters are theoretically validated to be favorable, exhibit chemical stability and strong aromaticity, and they may be targeted in future experiments to open a new area of coordination chemistry.
2. Computational details
The geometries of the planar wheel-type M©BnHn2−/−/0 and the bowl-type MBnHn0/+ (M = Cr, Mn and Fe for wheel-type, M = Cr and Mn for bowl-type; n = 6 and 7) were optimized using TPSSh29/6-311+G(3df,p), PBE30/6-311+G(3df,p) and BP86 (ref. 31–33)/6-311+G(3df,p) levels and imaginary frequencies were checked at the same level. Wiberg bond indices (WBI)34 were computed at BP86/6-311+G(3df,p) level using natural bond orbital (NBO)35 to gain insight into the bonding pattern of them. In order to assess the aromatic character, nucleus-independent chemical shifts (NICS),36 proposed by Schleyer and co-workers, were calculated with the ghost atom lying 1 Å (NICS(1)) above and below the M atom. The NICS(1) values were performed at BP86/6-311+G(3df,p) level with the gauge including atomic orbital (GIAO)37 method. The binding energy (Eb) between the M2−/−/0 and BnHn is defined as Eb = E(M©BnHn2−/−/0) − E(M2−/−/0) − E(BnHn). The vertical electron affinity (VEA) and vertical ionization potential (VIP) of M©BnHn2−/−/0 are defined as the total energy difference between M©BnHn2−/−/0 ± e (+for VEA and − for VIP) and M©BnHn2−/−/0 clusters at the optimized geometry of M©BnHn2−/−/0. The Eb, VEA and VIP of the bowl-type clusters can be made by analogy with those of the wheel-type ones. All calculations were performed using the GAUSSIAN 09 (ref. 38) program packages. The dimensional plots of molecular configurations and orbitals were generated with the GaussView program.393. Results and discussion
3.1 Geometry structure and energies
The optimized geometries of singlet M©BnHn2−/−/0 and singlet MBnHn0/+are shown in Fig. 1 and 2. And their geometry parameters are listed in Table 1. Since the geometry parameters obtained at the three computation levels are similar, the results shown in Fig. 1 and 2 and Table 1 are only from BP86/6-311+G(3df,p) level. The structures shown in Fig. 1 and 2 are all identified to be true local minima by frequency check. As can be seen from Fig. 1A, adding 2e to the bowl-type CrB6H6 cannot achieve the planar wheel-type D6h Cr©B6H62− which is a transition state with one imaginary frequency that leads to the C6v bowl-type CrB6H62−. However, it is interesting that the D6h Cr©B6H62− can be stabilized by two Li+ cations coordinated above and below the Cr atom (see Fig. 1C). In order to compare with C6v bowl-type CrB6H6, the D6h Cr©B6H62− that comes from Li2Cr©B6H62− will be used in the following studies. Besides CrB6H6, MnB6H6+, CrB7H7 and MnB7H7+ can all attain the planar wheel-type structures with high D6h and D7h symmetries by adding 2e into them. And the neutral clusters, Fe©B6H6 and Fe©B7H7 are all the planar wheel-type structures with high D6h and D7h symmetries.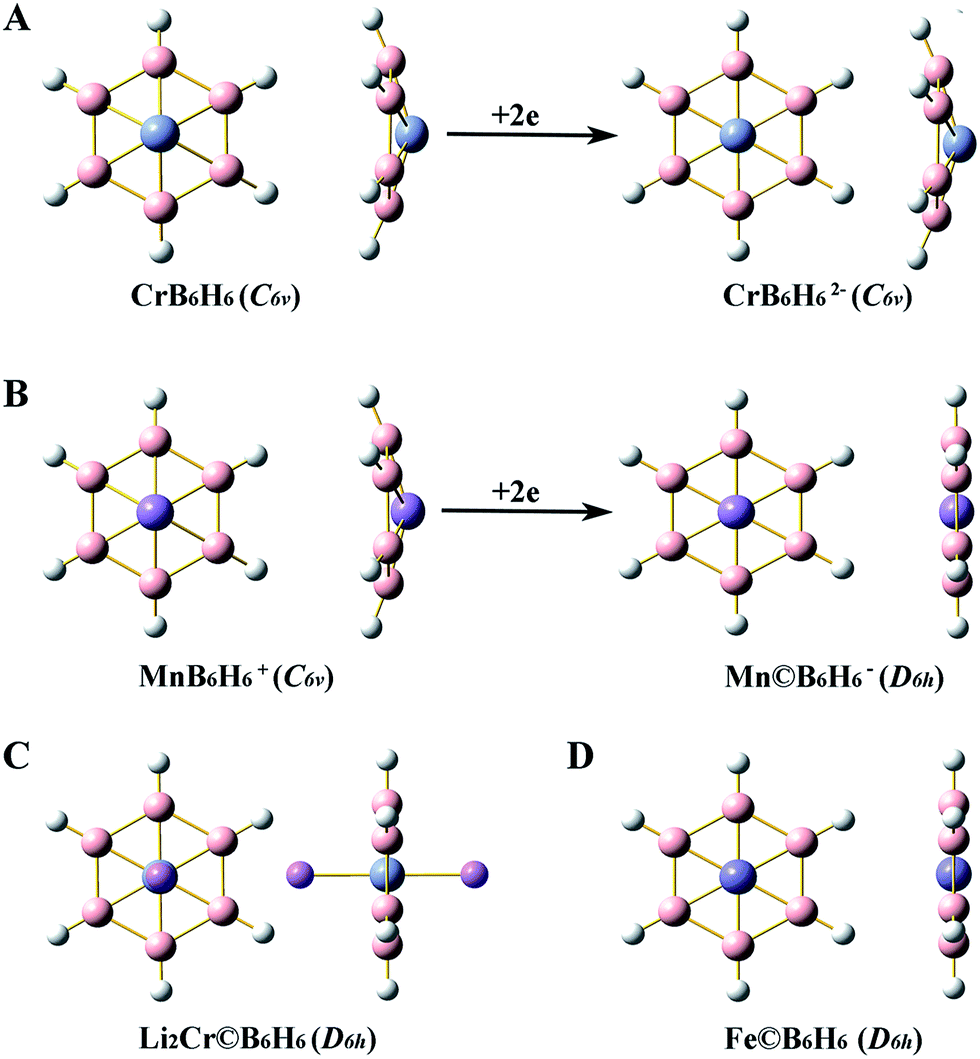 | ||
| Fig. 1 Optimized geometries of the singlet bowl-type and the singlet planar wheel-type clusters with hexagon boron ring at BP86/6-311+G(3df,p). H atoms are white and B atoms are pink. | ||
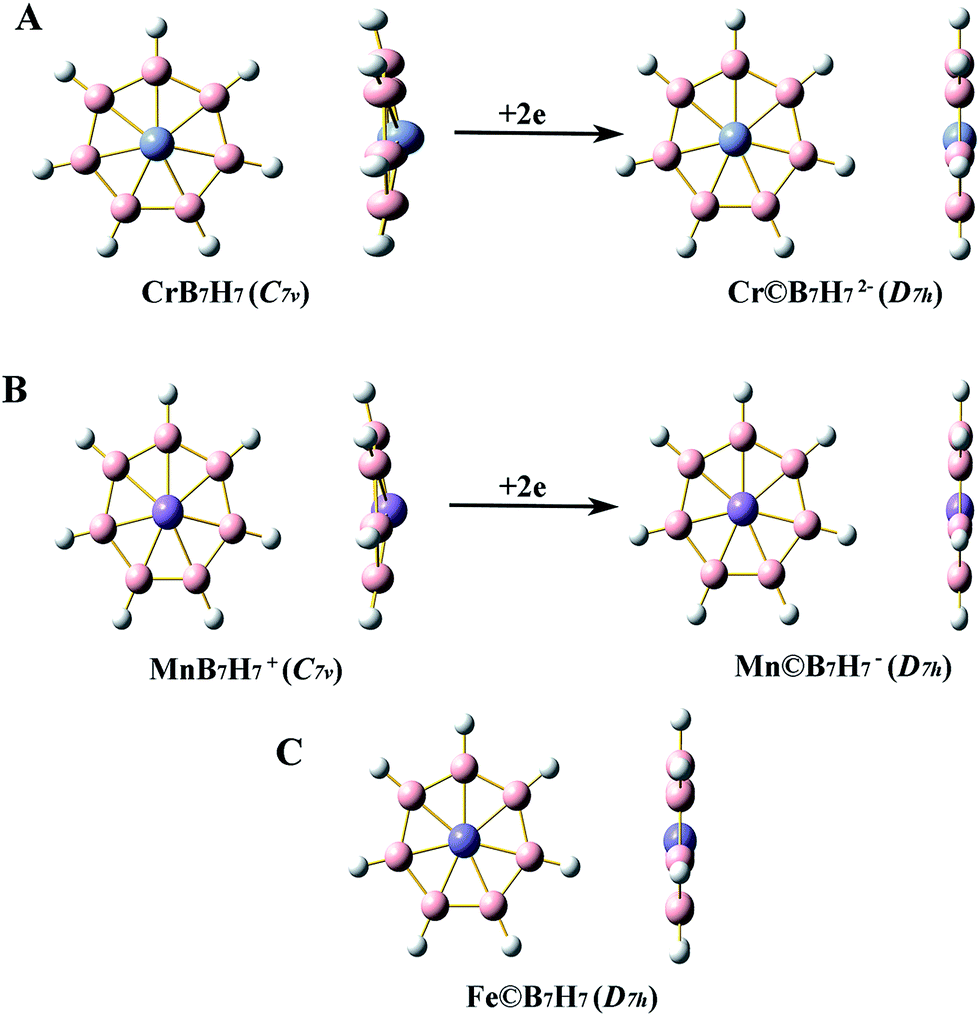 | ||
| Fig. 2 Optimized geometries of the singlet bowl-type and the singlet planar wheel-type clusters with heptagon boron ring at BP86/6-311+G(3d`). H atoms are white and B atoms are pink. | ||
| ∠BMB | RM–B | RB–B | RB–H | WBIM–B | WBIB–B | WBIM | WBIB | QM | QB | |
|---|---|---|---|---|---|---|---|---|---|---|
| a The average NBO charge for B atom. | ||||||||||
| Cr©B6H62− (D6h) | 60.0° | 1.887 | 1.887 | 1.199 | 1.01 | 0.64 | 6.16 | 3.57 | −1.57 | 0.26 |
| CrB6H6 (C6v) | 56.5° | 1.905 | 1.804 | 1.196 | 0.99 | 0.61 | 6.03 | 3.47 | −1.33 | 0.20 |
| Mn©B6H6− (D6h) | 60.0° | 1.851 | 1.851 | 1.204 | 0.92 | 0.65 | 5.62 | 3.53 | −2.00 | 0.21 |
| MnB6H6+ (C6v) | 57.2° | 1.890 | 1.809 | 1.193 | 0.89 | 0.57 | 5.46 | 3.30 | −1.29 | 0.32 |
| Fe©B6H6 (D6h) | 60.0° | 1.832 | 1.832 | 1.194 | 0.85 | 0.61 | 5.20 | 3.43 | −1.72 | 0.29 |
| Cr©B7H72− (D7h) | 51.4° | 1.988 | 1.725 | 1.212 | 0.84 | 0.78 | 5.97 | 3.64 | −2.18 | 0.1a |
| CrB7H7 (C7v) | 50.8° | 1.996 | 1.713 | 1.197 | 0.81 | 0.72 | 5.78 | 3.49 | −1.05 | 0.11 |
| Mn©B7H7− (D7h) | 51.4° | 1.958 | 1.699 | 1.205 | 0.76 | 0.78 | 5.42 | 3.59 | −1.64 | 0.13a |
| MnB7H7+ (C7v) | 50.6° | 1.995 | 1.706 | 1.196 | 0.74 | 0.70 | 5.31 | 3.36 | −0.95 | 0.19 |
| Fe©B7H7 (D7h) | 51.4° | 1.944 | 1.687 | 1.198 | 0.68 | 0.75 | 4.97 | 3.48 | −1.56 | 0.25a |
From the point of view of geometry parameters, it can be said that in general the B–M–B angles, the bond lengths of B–B and B–H bonds of the planar wheel-type clusters are larger than those of the bowl-type ones. In contrast the variation trend of the bond lengths of M–B bond is just the opposite. These results indicate that the interaction between the M atom and the boron ring in the planar wheel-type clusters is stronger than that in the bowl-type ones. As can be seen from Table 1, all B–B bond lengths are significantly longer than the typical B–B bond distances (1.593 Å) reported in the CB62−,40 but are still in the single-bond range of 1.706 to 1.859 Å (ref. 41), except for the longest bond length (1.887 Å) in Cr©B6H62−, which indicates very weak B–B interactions. In contrast the M–B bond distances (RCr–B = 1.887–1.996 Å, RMn–B = 1.851–1.995 Å and RFe–B = 1.832–1.944 Å) are notably shorter than the M–B covalent radii sums42 (2.25, 2.23 and 2.16 Å for M = Cr, Mn and Fe, respectively), which suggests the existence of robust interactions between the peripheral boron ring and the central M atom.
To compare the energy relationships of the planar wheel-type M©BnHn2−/−/0 with different spin multiplicity, we calculated the single point energies of the corresponding triplet, quintet and septet frames for these planar wheel-type clusters at BP86/6-311+G(3df,p) level. The relative energies based on the singlet are listed in Table 2. We found that the energy of the singlet is the lowest, except for Cr©B6H62− and Cr©B7H72−, as the relative energy increases with an increase of the spin multiplicity. For Cr©B6H62− and Cr©B7H72−, the energy of the triplet is the lowest. However, further optimization shows that the triplet Cr©B6H62− is also a transition state as the singlet and the symmetry of the triplet Cr©B7H72− is reduced from D7h to C2v. For the sake of comparison, the structures of singlet M©BnHn2−/−/0 will be used in the following studies.
| Spin multiplicity | Cr©B6H62− (D6h) | Mn©B6H6− (D6h) | Fe©B6H6 (D6h) | Cr©B7H72− (D7h) | Mn©B7H7− (D7h) | Fe©B7H7 (D7h) |
|---|---|---|---|---|---|---|
| 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| 3 | −0.33 | 0.66 | 1.14 | −0.08 | 1.11 | 1.52 |
| 5 | 2.65 | 3.48 | 3.48 | 3.01 | 3.96 | 4.34 |
| 7 | 5.72 | 6.60 | 6.61 | 5.96 | 6.73 | 6.38 |
3.2 WBIs and NBO charges
The WBIs of the M–B and the B–B bonds, total WBIs of M and B atoms and the NBO charges for M and B atoms are also listed in Table 1. The Wiberg bond order analyses are consistent with the feature of the geometries. For example, the WBIB–B = 0.57–0.78 are about half of WBIB–B = 1.15 computed for D2d B2H4 at the same level, which indicates the strength of the B–B single bond is weak in our systems. The total WBIB = 3.30–3.64 is close to 4, which implies that the B atoms follow the octet rule in bonding nature. The WBIM–B = 0.74–1.01 and total WBIM = 4.97–6.16 indicate that the M atom interacts strongly with the B atom and the boron ring.As can be seen from Table 1, the M atoms serve as negatively charged centers while the signs of the (average) charges on B atoms are positive. H atoms in all clusters carry negligible charges are not shown in Table 1. The interesting charge distribution that seems contradictory to the electronegativity indicates that not only does M atom donate electrons to boron atoms, but also the boron atoms donate electrons back to the M atom, which is important for these clusters to achieve stable structures.
3.3 Stability
The total energy differences ΔE(M,n) = E(D6h M©BnHn2−/−) − E(C6v MBnHn0/+), where M = Cr and Mn, n = 6 and 7, are tabulated in Table 3. It is obvious that D6h M©BnHn2−/− is more stable than C6v MBnHn0/+ at the three different computation levels except for Cr©B6H62− due to electrostatic repulsion. However, concerning the thermodynamic stability of D6h Li2Cr©B6H6, the energy change of the following process CrB6H6 (C6v 1A1) + 2Li → Li2Cr©B6H6−/0/+ (D6h 1A1g) was calculated. With zero-point correction included, we find that the reaction is highly exothermic (ΔE = −4.33 eV), indicating that the desired product is favored thermodynamically.| E(Cr, 6) | E(Mn, 6) | E(Cr, 7) | E(Mn, 7) | |
|---|---|---|---|---|
| BP86 | 0.420 | −10.063 | −0.813 | −11.571 |
| PBE | 0.583 | −9.860 | −0.634 | −11.400 |
| TPSSh | 0.903 | −9.895 | −0.366 | −11.423 |
The HOMO–LUMO energy gap (ΔH–L), Eb, VIP and VEA of D6h M©BnHn2−/−/0 and C6v MBnHn0/+ are listed in Table 4. The results of CrB6H6 and CrB7H7 are comparable to those obtained at TPSSh/6-311G(d) level.28 We can find that the ΔH–L of M©BnHn2−/− is smaller than that of MBnHn0/+ except for Mn©B7H7−. Compared with the neutral clusters, the VIP of M©BnHn2−/− is small due to electrostatic repulsion. However, the Eb of M©BnHn2−/− is bigger than that of MBnHn0/+, exhibiting better chemical stability. Moreover, for the isoelectronic neutral clusters Fe©B6H6 and Fe©B7H7, the indices are all improved, which suggests they are highly stable and may be observed in experiment.
| H–L | Eb | VIP | VEA | |
|---|---|---|---|---|
| Cr©B6H62− (D6h) | 0.24 | 11.89 | 2.74 | — |
| CrB6H6 (C6v) | 1.86 | 10.40 | 8.85 | 2.30 |
| Mn©B6H6− (D6h) | 0.90 | 12.33 | 2.07 | — |
| MnB6H6+ (C6v) | 1.79 | 9.56 | — | 7.90 |
| Fe©B6H6 (D6h) | 1.36 | 10.31 | 8.22 | 1.69 |
| Cr©B7H72− (D7h) | 0.14 | 16.13 | 2.64 | — |
| CrB7H7 (C7v) | 1.42 | 12.07 | 9.35 | 3.27 |
| Mn©B7H7− (D7h) | 1.47 | 15.62 | 2.54 | — |
| MnB7H7+ (C7v) | 0.96 | 10.93 | — | 8.69 |
| Fe©B7H7 (D7h) | 1.77 | 12.78 | 8.43 | 1.68 |
3.4 Aromaticity and molecular orbital
According to a perfect planar structure, we can propose that the planar wheel-type M©BnHn2−/−/0 should possess aromaticity. NICS is a simple and efficient criterion to characterize aromatic nature. A large negative NICS above molecular plane implies the presence of diamagnetic ring currents. Fig. 3 plots the NICS(1)s of the planar wheel-type M©BnHn2−/−/0, the bowl-type MBnHn0/+ and D6h C6H6. As shown in Fig. 3, the NICS(1)s above and below the plane of M©BnHn2−/−/0 are equal, and are all negative, which can prove that the diamagnetic ring current effect, characteristic for aromticity, exists in M©BnHn2−/−/0. Moreover, we find that the planar M©BnHn2−/−/0 have very strong aromatic character since their NICS(1)s are more negative than C6H6. While the NICS(1)s above and below the plane of the bowl-type MBnHn0/+ are not equal. The NICS(1)s in the bowl are much more negative than the one outside the bowl. Especially, the NICS(1) outside the bowl is positive in CrB6H6. The results indicate that the aromaticity outside the bowl is much weaker than that in the bowl, or even does not exist. The difference of the NICS(1) between inside and outside the bowl is attributed to the distribution of a π electron that mainly locates inside the bowl (see HOMO − 2 and HOMO − 1 in Fig. 4A and C).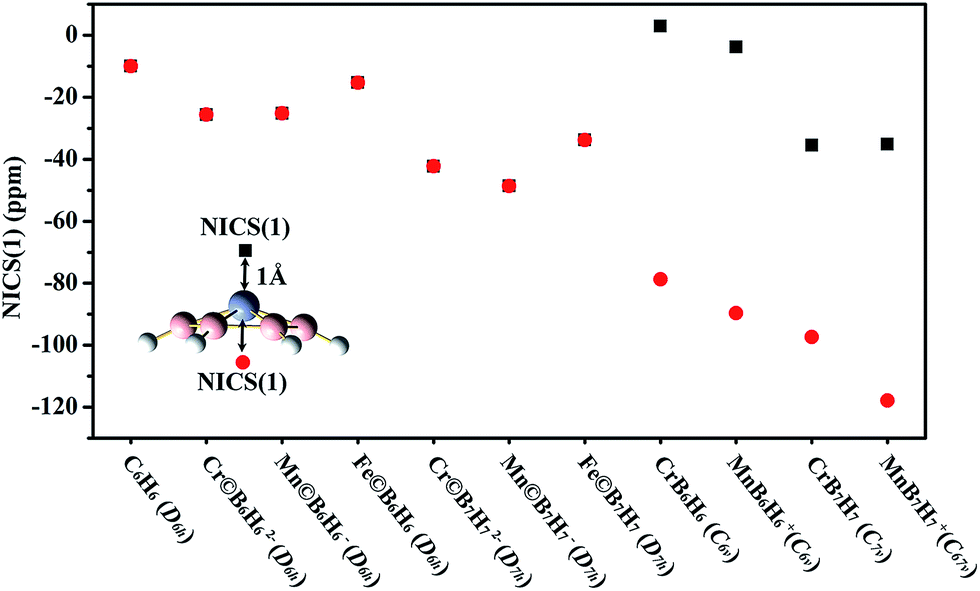 | ||
| Fig. 3 NICS(1)s of the singlet bowl-type and the singlet planar wheel-type clusters compared with those of D6h C6H6. | ||
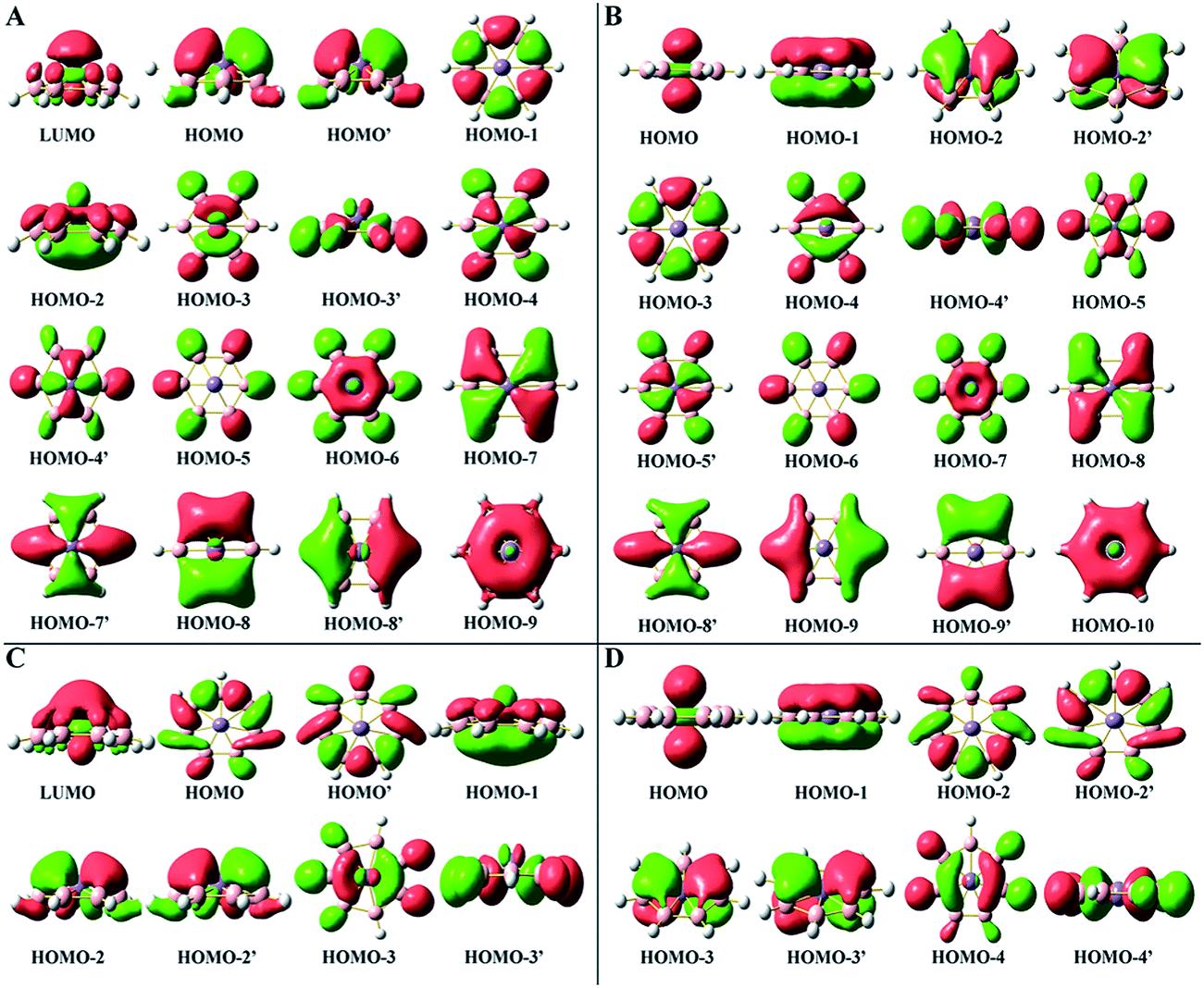 | ||
| Fig. 4 Molecular orbitals of C6v MnB6H6+ (A), D6h Mn©B6H6− (B), C7v MnB7H7+ (C) and D7h Mn©B7H7− (D) which are singlet. | ||
In order to better understand the perfectly planar wheel-type structure, we analyzed the molecular orbitals (MOs) of M©BnHn2−/−/0 and MBnHn0/+. The MOs of all isoelectronic clusters are very similar and the slight difference is the MOs order. Therefore, we mainly focus on the MOs of MnBnHn+ and Mn©BnHn− (n = 6 and 7). Density plots of the MOs are shown in Fig. 4. From the MnB6H6+ MOs shown in Fig. 4A, we can easily see that the degenerate HOMO and HOMO − 2 are the distorted π MOs that are responsible for the bowl-type structure. The reason is that the dz2 orbital of the center Mn atom involves in π MOs consisted of pz orbitals of boron ring. The strong interaction between them leads to the formation of LUMO and HOMO − 2. Thus, Mn dxz and dyz orbitals do not completely delocalize over the pz orbitals of the B atoms (see the degenerate HOMO), and parts of them participate in the px and py orbitals of the B atoms (see the degenerate HOMO − 3 and HOMO − 8). The remaining MOs do not include Mn dz2, dxz and dyz orbitals. However, as can be seen from Fig. 4B that shows the MOs of Mn©B6H6−, the additional two electrons more than in MnB6H6+ occupy the HOMO and form the nonbonding lone pair on the dz2 orbital of Mn atom. As a result, Mn dz2 orbital takes no part in the π MOs of the boron ring (see the HOMO − 1) and the Mn dxz and dyz orbitals completely extend to the pz orbitals of the B atoms (see the degenerate HOMO − 2), which makes the HOMO − 1 and the degenerate HOMO − 2 become fully normal π MOs that exhibit the typical pattern of aromatic molecule, benzene. Therefore, Mn©B6H6− with six π electrons is considered to be aromatic, conforming to the (4n + 2) Huckel rule. It is the π aromaticity that is responsible for the perfectly planar wheel-type structure. The important MOs that can determine the structures of MnB7H7+ and Mn©B7H7− are exhibited in Fig. 4C and D, which are similar to the previously mentioned case.
4. Conclusion
In summary, we adopted a new “electronic” strategy that is maintaining the lone pair on the center M atom, which is in contrast to the Hoffmann’s “electronic” strategy that is delocalizing them, to achieve planar wheel-type M©BnHn2−/−/0 clusters (M = Cr, Mn and Fe; n = 6 and 7). The lone pair occupies the dz2 orbital and makes it take no part in the π MOs, which is important for the planar wheel-type structures. The chemical stability of planar wheel-type structures is comparable with that of the bowl-type structures. These planar wheel-type M©BnHn2−/−/0 clusters deserve experimental confirmation because they would open up a new area in coordination chemistry for planar hexa- and hepta-coordinate transition metal.Acknowledgements
This work has been supported by the National Natural Science Foundation of China (nos 21201022 and 11464035), the Specialized Research Fund for the Doctoral Program of Higher Education (New Teachers, no. 20122216120001), the Natural Science Foundation of Jilin Province, China (no. 20120435, 20130204033GX, 20140101052JC, 20140204017GX and 20140101098JC).References
- R. Hoffmann, R. W. Alder and C. F. Wilcox Jr, Planar tetracoordinate carbon, J. Am. Chem. Soc., 1970, 92, 4992–4993 CrossRef CAS.
- K. Sorger and P. v. R. Schleyer, Planar and inherently non-tetrahedral tetracoordinate carbon: a status report, J. Mol. Struct.: THEOCHEM, 1995, 338, 317–346 CrossRef.
- X. Li, L. S. Wang, A. I. Boldyrev and J. Simons, Tetracoordinated Planar Carbon in the Al4C− Anion. A Combined Photoelectron Spectroscopy and ab Initio Study, J. Am. Chem. Soc., 1999, 121, 6033–6038 CrossRef CAS.
- L. S. Wang, A. I. Boldyrev, X. Li and J. Simons, Experimental Observation of Pentaatomic Tetracoordinate Planar Carbon-Containing Molecules, J. Am. Chem. Soc., 2000, 122, 7681–7687 CrossRef CAS.
- X. Li, H. F. Zhang, L. S. Wang, G. D. Geske and A. I. Boldyrev, Pentaatomic Tetracoordinate Planar Carbon, [CAl4]2−: A New Structural Unit and Its Salt Complexes, Angew. Chem., Int. Ed., 2000, 39, 3630–3633 CrossRef CAS.
- S. D. Li, G. M. Ren, C. Q. Miao and Z. H. Jin, M4H4X: Hydrometals (M = Cu, Ni) Containing Tetracoordinate Planar Nonmetals (X = B, C, N, O), Angew. Chem., Int. Ed., 2004, 43, 1371–1373 CrossRef CAS PubMed.
- Y. Pei, W. An, K. Ito, P. v. R. Schleyer and X. C. Zeng, Planar Pentacoordinate Carbon in CAl5+: A Global Minimum, J. Am. Chem. Soc., 2008, 130, 10394–10400 CrossRef CAS PubMed.
- K. Exner and P. v. R. Schleyer, Construction Principles of “Hyparenes”: Families of Molecules with Planar Pentacoordinate Carbons, Science, 2000, 290, 1937–1940 CrossRef CAS.
- Z. X. Wang and P. v. R. Schleyer, Planar Hexacoordinate Carbon: A Viable Possibility, Science, 2001, 292, 2465–2469 CrossRef CAS PubMed.
- H. J. Zhai, A. N. Alexandrova, K. A. Birch, A. I. Boldyrev and L. S. Wang, Hepta- and Octacoordinate Boron in Molecular Wheels of Eight- and Nine-Atom Boron Clusters: Observation and Confirmation, Angew. Chem., Int. Ed., 2003, 42, 6004–6008 CrossRef CAS PubMed.
- S. D. Li, C. Q. Miao, J. C. Guo and G. M. Ren, Planar Tetra-, Penta-, Hexa-, Hepta-, and Octacoordinate Silicons:? A Universal Structural Pattern, J. Am. Chem. Soc., 2004, 126, 16227–16231 CrossRef CAS PubMed.
- M. Lein, J. Frunzke and G. Frenking, A Novel Class of Aromatic Compounds: Metal-Centered Planar Cations [Fe(Sb5)]+ and [Fe(Bi5)]+, Angew. Chem., Int. Ed., 2003, 42, 1303–1306 CrossRef CAS PubMed.
- X. Li, B. Kiran, L. F. Cui and L. S. Wang, Magnetic Properties in Transition-Metal-Doped Gold Clusters: M@Au6 (M = Ti, V, Cr), Phys. Rev. Lett., 2005, 95, 253401 CrossRef.
- H. Tanaka, S. Neukermans, E. Janssens, R. Silverans and P. Lievens, Aromaticity of the Bimetallic Au5Zn+ Cluster, J. Am. Chem. Soc., 2003, 125, 2862–2863 CrossRef CAS PubMed.
- T. Höltzl, E. Janssens, N. Veldeman, T. Veszprémi, P. Lievens and M. T. Nguyen, The Cu7Sc Cluster is a Stable σ-Aromatic Seven-Membered Ring, ChemPhysChem, 2008, 9, 833–838 CrossRef PubMed.
- Z. Pu, K. Ito, P. v. R. Schleyer and Q. S. Li, Planar Hepta-, Octa-, Nona-, and Decacoordinate First Row d-Block Metals Enclosed by Boron Rings, Inorg. Chem., 2009, 48, 10679–10686 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, A. P. Sergeeva, W. L. Li, L. S. Wang and A. I. Boldyrev, Experimental and computational evidence of octa- and nona-coordinated planar iron-doped boron clusters: Fe©B8− and Fe©B9−, J. Organomet. Chem., 2012, 148, 721–722 Search PubMed.
- C. Romanescu, T. R. Galeev, W. L. Li, A. I. Boldyrev and L. S. Wang, Aromatic Metal-Centered Monocyclic Boron Rings: Co©B8− and Ru©B9−, Angew. Chem., Int. Ed., 2011, 50, 9334–9336 CrossRef CAS PubMed.
- W. L. Li, C. Romanescu, Z. A. Piazza and L. S. Wang, Geometrical requirements for transition-metal-centered aromatic boron wheels: the case of VB10−, Phys. Chem. Chem. Phys., 2012, 14, 13663–13669 RSC.
- W. L. Li, C. Romanescu, T. R. Galeev, Z. A. Piazza, A. I. Boldyrev and L. S. Wang, Transition-Metal-Centered Nine-Membered Boron Rings: M©B9 and M©B9− (M = Rh, Ir), J. Am. Chem. Soc., 2012, 134, 165–168 CrossRef CAS PubMed.
- T. R. Galeev, C. Romanescu, W. L. Li, L. S. Wang and A. I. Boldyrev, Observation of the Highest Coordination Number in Planar Species: Decacoordinated Ta©B10− and Nb©B10− Anions, Angew. Chem., Int. Ed., 2012, 51, 2101–2105 CrossRef CAS PubMed.
- C. Romanescu, T. R. Galeev, L. L. Wei, A. I. Boldyrev and L. S. Wang, Geometric and electronic factors in the rational design of transition-metal-centered boron molecular wheels, J. Chem. Phys., 2013, 138, 134315 CrossRef PubMed.
- W. L. Li, C. Romanescu, T. Jian and L. S. Wang, Elongation of Planar Boron Clusters by Hydrogenation: Boron Analogues of Polyenes, J. Am. Chem. Soc., 2012, 134, 13228–13231 CrossRef CAS PubMed.
- D. Z. Li, H. G. Lu and S. D. Li, Planar π-aromatic C3h B6H3+ and π-antiaromatic C2h B8H2: boron hydride analogues of D3h C3H3+ and D2h C4H4, J. Mol. Model., 2012, 18, 3161–3167 CrossRef CAS PubMed.
- D. Z. Li, Q. Chen, Y. B. Wu, H. G. Lu and S. D. Li, Double-chain planar D2h B4H2, C2h B8H2, and C2h B12H2: conjugated aromatic borenes, Phys. Chem. Chem. Phys., 2012, 14, 14769–14774 RSC.
- Q. Chen and S. D. Li, Aromatic B16H6: A Neutral Boron Hydride Analogue of Naphthalene, J. Cluster Sci., 2011, 22, 513–523 CrossRef CAS.
- H. L. Yu, R. L. Sang and Y. Y. Wu, Structure and Aromaticity of B6H5+ Cation: A Novel Borhydride System Containing Planar Pentacoordinated Boron, J. Phys. Chem. A, 2009, 113, 3382–3386 CrossRef CAS PubMed.
- L. F. Li, C. Xu, B. K. Jin and L. J. Cheng, Benzene analogues of (quasi) planar M@BnHn compounds (M = V−, Cr, Mn+): a theoretical investigation, J. Chem. Phys., 2013, 139, 174310 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Climbing the Density Functional Ladder: Nonempirical Meta-Generalized Gradient Approximation Designed for Molecules and Solids, Phys. Rev. Lett., 2003, 91, 146401–146404 CrossRef.
- J. P. Perdew, K. Burke and M. Enzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. D. Becke, Density-functional exchange-energy approximation with correct asymptotic behavior, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- J. P. Perdrew, Density-functional approximation for the correlation energy of the inhomogeneous electron gas, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 34, 7406–7408 CrossRef.
- K. P. Jensen, B. O. Roos and U. Ryde, Performance of density functionals for first row transition metal systems, J. Chem. Phys., 2007, 126, 014103 CrossRef PubMed.
- K. B. Wiberg, Application of the pople-santry-segal CNDO method to the cyclopropylcarbinyl and cyclobutyl cation and to bicyclobutane, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- J. E. Carpenter and F. Weinhold, Analysis of the geometry of the hydroxymethyl radical by the “different hybrids for different spins” natural bond orbital procedure, J. Mol. Struct., 1988, 169, 41–62 CrossRef.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. v. E. Hommes, Nucleus-Independent Chemical Shifts: A Simple and Efficient Aromaticity Probe, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS.
- R. Ditchfield, Self-consistent perturbation theory of diamagnetism I. A gauge-invariant LCAO method forN.M.R. chemical shifts, Mol. Phys., 1974, 27, 789–807 CrossRef CAS.
- M. J. T. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, GAUSSIAN 09, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Package GaussView 5.0.9, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- R. Islas, T. Heine, K. Ito, P. v. R. Schleyer and G. Merino, Boron Rings Enclosing Planar Hypercoordinate Group 14 Elements, J. Am. Chem. Soc., 2007, 129, 14767–14774 CrossRef CAS PubMed.
- P. P. Power, Bonding and the Lone Pair Effect in Multiple Bonds between Heavier Main Group Elements, Chem. Rev., 1999, 99, 3463–3503 CrossRef CAS PubMed.
- B. Cordero, V. Gomez, A. E. Platero-Prats, M. Reves, J. Echeverrıa, E. Cremades, F. Barragan and S. Alvarez, Covalent radii revisited, Dalton Trans., 2008, 2832–2838 RSC.
| This journal is © The Royal Society of Chemistry 2015 |