
Development of a novel and efficient H2O2 sensor by simple modification of a screen printed Au electrode with Ru nanoparticle loaded functionalized mesoporous SBA15†
Subhenjit Hazraa,
Hrishikesh Joshia,
Barun Kumar Ghosha,
Asif Ahmedb,
Timothy Gibsonb,
Paul Millnerb and
Narendra Nath Ghosh*a
aNano-Materials Lab, Department of Chemistry, Birla Institute of Technology and Science, Pilani K. K. Birla Goa Campus, Zuarinagar, Goa 403726, India. E-mail: naren70@yahoo.com; Fax: +91 832 2557033; Tel: +91 832 2580318
bSchool of Biomedical Sciences, Faculty of Biological Sciences, University of Leeds, Leeds, UK
First published on 8th April 2015
Abstract
A novel and efficient electrochemical sensor has been developed to quantitatively measure H2O2 concentration by cyclic voltammetry. The sensor was prepared by modifying screen printed gold electrodes by ruthenium nanoparticle (Ru nanoparticle) loaded thiol functionalized mesoporous SBA15 (Ru@SBA15-SH) which was prepared by three simple steps. During measurement H2O2 electrochemically interacted with Ru nanoparticles and was channelled appropriately through the mesoporous structure of SBA15. The developed sensor showed a wide detection range with high sensitivity, durability and reproducibility. Furthermore, a very low limit of detection was reported by the sensor (0.42 μM (∼0.0142 ppm)), which was much lower than the permissible exposure limit.
1. Introduction
Detection of hydrogen peroxide (H2O2) at a very low concentration level, ppm and below, has become of immense interest to the scientist and technologists. H2O2 has plethora of applications in pharmaceutical, clinical, environmental, mining, textile and food manufacturing industries.1,2 In many industries very high concentrations of H2O2 (greater than 10% w/v) in aqueous solutions are commonly used, which are very oxidizing and corrosive upon contact and cause severe inflammation to various organs of the body such as mucus membranes, gastrointestinal mucosa, skin and eyes.3,4 Moreover, in living organisms, besides its well-known cytotoxic effects, H2O2 also plays a critical role in regulating diverse biological processes as a signaling molecule in apoptosis, immune cell activation, stomatal closure, vascular remodelling and root growth processes.5,6 Formation of H2O2 also occurs as a side product from some classic biochemical reactions catalyzed by enzymes such as glucose oxidase (GOx), alcohol oxidase (AlOx), lactate oxidase (Lox) cholesterol oxidase (ChoOx), etc.7According to the occupational health and safety administration's norm the permissible exposure limit for inhalation of H2O2 vapour is 1 ppm and concentration levels over 7 ppm are known to cause lung irritation.8 Therefore, in the academic as well as industrial point of view investigations on H2O2 detection are of practical significance. Although, conventional techniques are available for hydrogen peroxide determination such as titrimetric,9 spectrophotometric,10–13 deviations in electrical conductivity,14 fluorometric,15–17 liquid chromatography,18 chemiluminescence19–21 and electrochemiluminescence22,23 but they all are complex in nature and in some cases expensive and time consuming.
Since H2O2 is an electroactive molecule, detection of H2O2 by using electrochemistry is the most attractive technique, owing to its cost-effectiveness, portability and instrumental simplicity.24 H2O2 can be detected via either oxidation or reduction at solid electrode surfaces. However, the major concern of electrochemistry is its slow kinetics and high over potential. This affects the sensing performance and may incur substantial interference from existing electroactive species, such as ascorbate, urate and bilirubin in real samples. Therefore, in search for an efficient H2O2 sensing electrode, a large range of materials, such as redox proteins, dyes, transition metals, metal oxides, metal phthalocyanines, metal porphyrins, redox polymers, carbon nanotubes, mesoporous materials etc., have been employed to detect peroxide over the years.25 Some of the mesoporous materials, such as SBA15, MCM-41 etc. possess a high specific surface area along with high porosity and long range ordering. These materials are often used as supports to host various catalytically active sites since their porous structures enhance the accessibility and mass transportation of the reactant molecules to the active centers.26 These materials have also shown promise to be used in various sensor applications due to their selectivity and ease of surface functionalization with various reactive groups.24–26
Over the years, several nanoparticle based electrochemical sensor have been developed for detection of H2O2 where different kinds of electrodes have been used. The glassy carbon (GC) electrode has been widely used in sensor development by modifying the electrode using nanomaterials such as platinum–gold nanoclusters on a graphene sheet,27 Ag nanoparticles28 and cadmium oxide nanoparticles embedded in multiwall carbon nanotubes (MWNT).29 Ferric hexacyanoferrate or Prussian blue (PB) is a well explored compound which catalyses H2O2 reduction because of the highly catalytically active reduced form of PB (Prussian white) and also due to the polycrystalline structure of PB.30,31 Several reports have suggested the use of Prussian blue (PB) modified electrodes coupled with carbon electrodes,32 carbon nanotubes/nanocomposites33 and even platinum/gold electrodes.34 The major drawback associated with PB is the lack of operational stability in neutral and alkaline solutions because the reduced form of PB, Prussian white, can be dissolved by hydroxide ions.35 Other metal hexacyanoferrates such as Cu, Ni, Co, Cr, V, Ru and Mn hexacyanoferrates can also be used for hydrogen peroxide sensing. These metal hexacyanoferrate based sensors have exhibited a lower capability for electrocatalytic reduction of H2O2 than PB based electrodes, but with greater electrochemical stability over a wide range of pH.8 Platinum36 and gold electrodes have also been used for sensor applications as both metals increase the response and sensitivity of the electrode.
In the last few years, a huge number of electrochemical devices have been successfully built using screen printing techniques.37 This technology offers multiple advantages including cost effectiveness, flexibility, process automation, reproducibility and usage of variety of materials. Screen printed electrodes can be coupled with the enzyme horseradish peroxidase (HRP) to detect H2O2 by using nanoparticles in chitosan matrix,38 labelling Au colloids immobilized on a gold electrode via a cysteamine monolayer with HRP39 or even by direct electron transfer between the screen printed electrode and HRP.40,41 Ruthenium complexes (such as K[Ru(EDTA-H)C1]·2H2O,42 Ru(III)phosphate43) were used in developing electrochemical sensors for H2O2, as these complexes could effectively perform electron transfer reaction with peroxide.44 Ruthenium(IV) oxide has also been known to interact with H2O2, and was used to study catalysis activity in plants.45 Apparently, the Ru hcp crystal structure of the nanoparticles adsorbs OHads as well as Oads readily, and hence, can successfully catalyze the H2O2 degradation in electrochemical system.46 However, the electrochemical sensors developed and reported until now, involve complex preparation protocols and suffers from poor response and sensitivity. Moreover, the sensors developed using HRP require specific conditions of pH, buffer and optimal temperature for effective detection and reproducibility. Furthermore, in many of the reported sensors the preparation method for the nanomaterials are quite complex.
In the present investigation, we have adopted a strategy to construct a composite material by immobilizing metal nanoparticles (here Ru nanoparticle) within the porous structure of mesoporous silica (SBA15) so that we can exploit both the advantages offered by nanoparticle as well as mesoporous silica. The preparation of these nanoparticles involves two simple post treatments of SBA15. In the first step Ru nanoparticles were incorporated in the silica matrix and second step involved thiol functionalization of the Ru nanoparticle incorporated SBA15. After functionalization of Ru@SBA15, the nanocomposites (Ru@SBA15-SH) were attached to the surface of screen printed Au electrodes by a simple drop cast technique. We modified the screen printed gold electrode (SPE) as it serves as a good and cost effective mediator for electron transfer. The synthesized material was characterized by BET, XRD, FTIR and electron microscopy.
Here, we report development of an efficient, sensitive and robust hydrogen peroxide sensor with a simple preparation technique. The functionalized materials were easily deposited on the gold electrodes as the thiol groups present on surface interacted effectively with the gold surface. A thorough electrochemical study was conducted to establish linearity and sensitivity of current response, reliability of sensor, durability of electrode and also to determine the limit of detection.
2. Experimental section
2.1 Materials
Tetraethyl orthosilicate (TEOS), conc. hydrochloric acid (HCl), EO20PO70EO20 (P123), ruthenium chloride hydrate (RuCl3·xH2O), and toluene were purchased from Merck, India. Custom made screen-printed gold electrodes (circular working electrodes, CX2223AT) were supplied by Drop Sens S.L. (Oviedo, Spain). These electrodes consist of ceramic base with Ag/AgCl reference and Au counter electrode on same chip. H2O2 (30% w/v solution) and (3-mercaptopropyl)trimethoxysilane (MPTMS) were purchased from Sigma Aldrich. Deionized water and 0.1 M PBS buffer, pH of 7.0 were used throughout the experiments.2.2 Synthesis of SBA15 (SBA15)
Mesoporous silicate SBA15 was synthesized using a liquid crystal templating method using Pluronic P123, tetraethyl orthosilicate and concentrated hydrochloric acid.47 In a typical synthesis of SBA15, 3.3 g of Pluronic P123 was dissolved in 101 g of water with 8 g of concentrated hydrochloric acid. Tte mixture was stirred until the formation of a clear solution. Then, 6.93 g of tetraethyl orthosilicate was added to this solution and stirred for 4 h. The resulting mixture was then transferred to a Teflon bottle and heated at 90 °C for 24 h to allow condensation. The precursor formed was then filtered and washed with 150 ml of distilled water and dried in the oven at 100 °C. It was then calcined at 550 °C for 3 h in an air atmosphere to obtain SBA15.2.3 Synthesis of ruthenium nanoparticle incorporated SBA15 (Ru@SBA15)
Ru nanoparticle loaded SBA15 samples (10% w/w Ru) were synthesized. Here, RuCl3·xH2O was reduced by sodium borohydrate (NaBH4) using a molar ratio of RuCl3·xH2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaBH4 of 1:5. In a typical synthesis, 450 mg of SBA15 was soaked overnight with 125 mg RuCl3·xH2O in 12 ml of water. To this mixture 340 mg of NaBH4 dissolved in 10 ml of water was slowly added and stirred for 8 h. Finally, the solution was filtered using Whatman-41 filter paper and product was vacuum dried.
:NaBH4 of 1:5. In a typical synthesis, 450 mg of SBA15 was soaked overnight with 125 mg RuCl3·xH2O in 12 ml of water. To this mixture 340 mg of NaBH4 dissolved in 10 ml of water was slowly added and stirred for 8 h. Finally, the solution was filtered using Whatman-41 filter paper and product was vacuum dried.
2.4 Synthesis of thiol functionalized Ru@SBA15 (Ru@SBA15-SH)
The synthesized Ru@SBA15 was thiol functionalized using (3-mercaptopropyl)trimethoxysilane (MPTMS). Here, 100 mg of Ru@SBA15 was dispersed in 10 ml of dry toluene and 500 μl of MPTMS was added to it. The reaction mixture was then refluxed under a nitrogen atmosphere for 8 h. Then the thiol fuctionalised Ru@SBA15 was filtered using Whatman-41 filter paper and thoroughly washed with dry toluene to remove excess MPTMS. Finally synthesized particles were dried under vacuum. After thiol functionalization, 1 g Ru@SBA15 resulted in the formation of 1.4 g Ru@SBA15-SH. A similar synthesis procedure was followed to synthesize a control material designated as SBA15-SH by using pure SBA15 instead of Ru@SBA15 to study the effect of Ru nanoparticle on peroxide detection.2.5 Electrode preparation
The electrode surface was cleaned by washing the surface with deionized water. 1 mg of thiol functionalized ruthenium embedded SBA15 (Ru@SBA15-SH) was dissolved in 100 μl of deionized water and sonicated for 5 min. ∼5 μl of the solution was deposited onto the gold surface of the electrode and kept in a moist atmosphere for 4 h to immobilize the particles on the surface. The electrode was then gently washed with a stream of deionized water and then dried by gently blowing nitrogen over it. The overall protocol for the preparation of the electrode is given in Scheme 1. A control electrode was also prepared by a similar procedure but by using SBA-SH instead of Ru@SBA15-SH. | ||
| Scheme 1 Schematic representation of preparation of Ru@SBA15-SH based sensor. | ||
2.6 Characterization
Thermogravimetric analysis (TGA) was carried out for Ru@SBA15 and Ru@SBA15-SH using a DTG-60 (Shimaduzu, Japan) in an air flow at a heating rate of 10 °C min−1 between 30 °C to 800 °C. Platinum sample pans were used for TGA. Room temperature X-ray diffraction spectra of the samples were recorded by using a powder X-ray diffractometer (Mini Flex II, Rigaku, Japan) with Cu Kα (λ = 0.15405 nm) radiation. Transmission electron microscopic (HRTEM) (JEOL JEM 1400, Japan) images of samples were used to analyze the pores and size of the synthesized nanoparticles. The morphology of the nanoparticles, deposited on the electrode surface, was studied using scanning electron microscopy (SEM) (JSM-6360LV, JEOL, Japan) using an accelerating voltage of 15 kV. Energy-dispersive X-ray spectroscopy (EDS) spectra was recorded using JEOL JSM-5800LV scanning microscopy (JEOL, Japan). Nitrogen adsorption–desorption isotherms, surface area, pore diameter, pore volume were obtained with a surface area and porosity analyzer (Micromeritics Tristar 3000, USA). FTIR spectroscopy was carried out using FTIR-IR infinity-101660, (Shimadzu, Japan). All of the samples were milled with spectroscopic grade potassium bromide (KBr, Merck), and the pellet of mixture was pressed into a disc and placed in solid cell and scanned in the range 4000–400 cm−1. Electrochemical studies were performed using Eco Chemie B V Autolab electrochemical workstation using GPES software for cyclic voltammetry study and FRA software for the impedance study. All electrochemical experiments were performed in open air condition. We have reported here the highly reproducible cyclic voltammogram which is average of two successive measurements. The limit of detection (LOD) was calculated as 3Sa/b where Sa denotes the standard deviation of the intercept and b is the slope for the linear fit ([I − Io] = a + b[H2O2]) where Io is current value at [H2O2] = 0 and signal/noise ratio is 3.48–513. Results and discussion
3.1 Physical characterization of materials
X-ray diffractograms of the synthesized materials, e.g. SBA15, Ru@SBA15 and Ru@SBA15-SH are shown in Fig. 1. In the XRD patterns for Ru@SBA15 and Ru@SBA15-SH samples, diffraction peak at 2θ = 43.42° corresponding to (101) diffraction plane of hcp Ru [JCPDS card no. 65-1863] was observed along with the broad peak of SBA15. XRD patterns of Ru@SBA15 and Ru@SBA15-SH also showed diffraction peaks at 2θ = 38.38°, 58.3°, 69.44°, 78.38° corresponding to (100), (102), (110), and (103) diffraction planes of hcp Ru [JCPDS card no. 65-1863] (Fig. S1(A) and (B)†).52–54 However, these peaks were found to be very weak, because of the fact that Ru nanoparticles are embedded within the porous matrix of SBA15. Crystallite size of Ru nanoparticles was calculated using Schrrer's equation and was found to be ∼8 nm.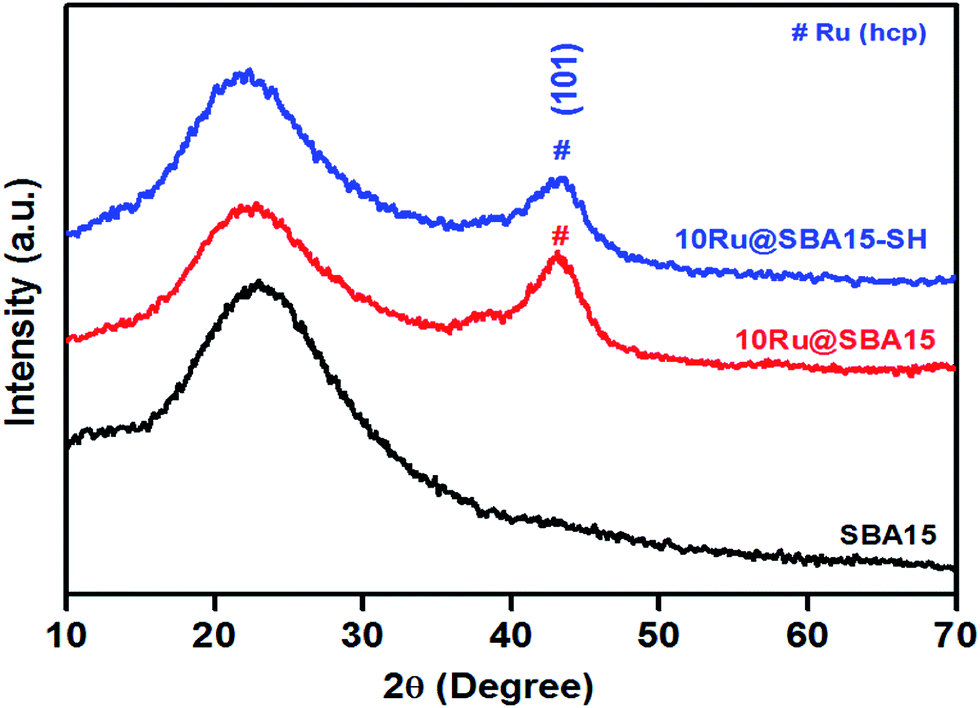 | ||
| Fig. 1 Wide angle XRD spectra of SBA15, Ru@SBA15 and Ru@SBA15-SH. | ||
TG analysis of Ru@SBA15 and Ru@SBA15-SH confirmed the functionalization of SBA15 by MPTMS (Fig. 2). In the thermogram of Ru@SBA15-SH, ∼15% weight loss was observed in the temperature range 150–550 °C, whereas no significant weight loss was observed in the case of Ru@SBA15. This weight loss might be due to the oxidative thermal decomposition of MPTMS which was present in Ru@SBA15-SH. This weight loss also indicated that, during thiol functionalization ∼0.33 mole MPTMS is attached with 1 mole of SBA15, which matches with the theoretically calculated amount of thiol functionalization in Ru@SBA15-SH (refer Section 2.4).
 | ||
| Fig. 2 TGA thermogram of Ru@SBA15-SH and Ru@SBA15. | ||
FTIR spectra of Ru@SBA15 and Ru@SBA15-SH are shown in Fig. 3, which confirmed the thiol functionalization of the material. The broad intense peak at 3500–3300 cm−1 in Ru@SBA15 was assigned to the Si–OH group of SBA15. The intensity of this peak was found to be significantly decreased when Ru@SBA15 was functionalized by MPTMS to prepare Ru@SBA15-SH. This fact indicated the reaction of Si–OH of Ru@SBA15 with –Si(OMe)3 group of MPTMS. FTIR spectra of Ru@SBA15 also showed the presence of peaks at 2574 cm−1 and 2956 cm−1 which are characteristic peaks of –SH group and –CH of propyl group MPTMS.
 | ||
| Fig. 3 FTIR spectra of Ru@SBA15-SH and Ru@SBA15. Inset graph: FTIR spectra showing –SH peak. | ||
N2 adsorption–desorption analysis of pure SBA15 and Ru@SBA15 (Fig. 4) showed type IV isotherms with a H1 hysteresis loop, which is characteristic of mesoporous material having a regular pore structure. However, the BET surface area was found to be decreased due to the 10% (w/w) Ru nanoparticle loading in Ru@SBA15. It was also observed that the upper closure point of the hysteresis loop of Ru@SBA15 (at P/P0 = 0.95) appeared at a relatively higher value than that of pure SBA15 (at P/P0 = 0.78). This fact indicates that formation of Ru nanoparticles within the pores of SBA15 blocked some of the channels and caused partial strain and distortion in the pores. Pore volume was also found to be decreased from 1.01 m3 g−1 to 0.76 m3 g−1 due to Ru nanoparticle loading into the SBA15 matrix.
 | ||
| Fig. 4 N2 adsorption and desorption isotherm of SBA15 and Ru@SBA15. | ||
TEM micrographs of the synthesized materials are shown in Fig. 5(a–d). Regular hexagonal porous structure with long range ordering and long channels with pore diameters of 7–10 nm were observed for pure SBA15. Ru loaded samples, i.e. Ru@SBA15 and Ru@SBA15-SH, showed the presence of uniform spherical shaped Ru nanoparticles (∼8 nm) within and on the surface of the porous matrix of SBA15. EDS analysis of the samples also confirmed the presence of Ru in Ru@SBA15 (Fig. S2†). Another important observation is that even after Ru loading as well thiol functionalization of Ru@SBA15 the porous nature of SBA15 long channels remained intact. The highly porous and regular ordered structure of Ru@SBA15 should enhance rapid electron transfer to enzyme actives sites during electrochemical analysis.
 | ||
| Fig. 5 TEM micrographs of (a) pure SBA15, (b) SBA-SH, (c) Ru@SBA15 and (d) Ru@SBA15-SH. | ||
Surface morphology of bare Au electrode and electrode surface after deposition of Ru@SBA15-SH and SBA15-SH was investigated by SEM (Fig. 6(a) and (b)). The micrographs revealed the relatively smooth surface of the bare electrode before deposition of Ru@SBA15-SH.
 | ||
| Fig. 6 SEM micrographs of (a) bare electrode, (b) Ru@SBA15-SH deposited electrode. | ||
3.2 Electrochemical studies
Electrochemical impedance spectroscopy (EIS) yields substantial information about the modifications on the electrode surface. Fig. 7 shows the impedance data of bare Au electrode, control electrode (SBA15-SH deposited on Au SPE), sensor electrode (Ru@SBA15-SH deposited on Au SPE) in 10 mM (1:1 ratio) [Fe(CN)6]3−/4−. The higher Rct values of sensor and control electrodes than the bare electrode (in Nyquist plots) clearly indicate the deposition of particles on both (control and sensor) electrodes. The equivalent circuit of the electrochemical system55 is given in Fig. 7. High Rct value of the sensor electrode can be accounted for by the fact that mesoporous channels in SBA15 have been blocked by the Ru nanoparticles, thereby reducing the accessibility of the gold surface for direct electron charge transfer. Also the cyclic voltammogram of the sensor (Fig. 8) showed a large deviation in oxidation and reduction potential whereas the control sensor shows almost no deviation proving that incorporated Ru nanoparticles caused the deviations in the electrochemical properties. As Ru is electrochemically very active, hence due to the electrochemical interaction between ferri-ferrocyanide and Ru nanoparticles, which are present in sensor electrode (i.e. Ru@SBA15-SH modified Au electrode), the cyclic voltammetry showed higher peak for Ru@SBA15-SH deposited Au electrode (sensor electrode) in comparison with SBA15-SH modified Au electrode (control electrode) and bare Au electrode (Ru nanoparticles were not present in control electrode and bare electrode).
 | ||
| Fig. 7 Nyquist plots of bare electrode, SBA15-SH deposited on electrode and Ru@SBA15-SH deposited on electrode. Inset picture: equivalent electrochemical circuit. | ||
 | ||
| Fig. 8 Cyclic voltammogram of bare electrode, SBA15-SH deposited on electrode and Ru@SBA15-SH deposited on the electrode. | ||
Ru has been known to be involved in various redox couple reactions with H2O2,56,57 hence the Ru nanoparticles are expected to be involved in the charge transfer to detect peroxide quantitatively. To conduct quantitative measurement of H2O2 the potential range was set from −0.4 V to 0.75 V in 0.1 M PBS. Potential scan from −0.6 V to 1.5 V was also carried out, however, in the cyclic voltammogram extra peaks for oxidation were appeared after 1 V (Fig. S3(A)†). Moreover, formation of some bubbles near the electrode surface was observed when scan was conducted from −0.6 V to 1.5 V. This might be due to the oxidation of the Au electrode surface. Reproducibility of the results was also reduced when scans were conducted with this high potential range (Fig. S3(B)†). The nature of the voltammogram was also altered after the high potential scan (Fig. S3(B)†). Therefore, the potential range from −0.4 V to 0.75 V was set all the measurements. Fig. 9(A) shows a cyclic voltammogram in 0.1 M PBS (pH 7) with increasing concentrations of H2O2 onto the sensor electrode. It was observed that current at 0.75 V is directly proportional to the concentration of H2O2. Fig. 9(B) suggests that such an enhanced current response is absent in the case of bare and control electrodes, thereby proving that only Ru nanoparticles when interacted with H2O2 generate enhanced response. There have been reports demonstrating the use of only gold electrodes to sense H2O2; but the reported current signal is almost 60 times lower than this sensor.30 The range of detection for H2O2 of this Ru@SBA15-SH modified SPE sensor is shown in Fig. 10. This sensor showed a linear trend over the mM as well as μM concentration ranges. The linear trend was also followed by the control and bare electrodes but with a much less current. It is suspected that this was due to charge transfer between the gold electrode and peroxide.3 There was not much difference in current values between bare and control electrodes (Fig. 9(B)) which signifies that the deposited silica particles caused almost no change to the electrochemical system. Hence, in case of the sensor Ru nanoparticles were responsible for the enhanced current values. The highest current value appeared at the oxidation peak which supports the mechanism of the charge transfer between peroxide and Ru nanoparticles. Peroxide oxidizes Ru nanoparticles to Run+ which occurs at a potential of 0.75 V (pH 7 solution). The linearity of the sensor ranged from 2 × 10−6 M to 10−1 M. Above this concentration the peroxide solution became highly corrosive and prolonged exposure of the electrode in the solution degraded the surface of the sensor. Sensitivity of the current response for the developed sensor was 11.26 μA mM−1 calculated using an average of n = 10 signals each having a triplicate reading.
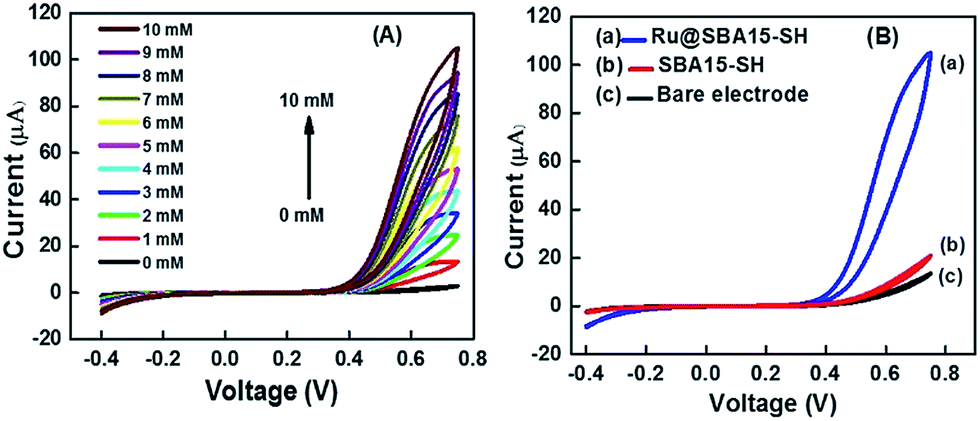 | ||
| Fig. 9 (A) Cyclic voltammograms of Ru@SBA15-SH electrode with varying concentration of H2O2, (B) cyclic voltammogram of (a) Ru@SBA15-SH, (b) SBA15-SH modified electrode and (c) bare electrode with [H2O2] = 10 mM. | ||
 | ||
| Fig. 10 Current value at (V = 0.75 V) vs. concentration over (A) mM range and (B) μM range. | ||
Fig. 11(A) shows a cyclic voltammogram of three electrodes (prepared by the same process) in 0.1 M PBS and interrogated with 10 mM H2O2. It was very clear that the current values were quite reproducible with no more than 6.2% relative standard deviations. Fig. 11(B) shows a plot of an electrode prepared on day 1 and tested with 10 mM and 5 mM of H2O2 on day 31 (∼1 month apart). The plot shows almost similar response on both the days for 5 mM as well as 10 mM which indicates the durability of the sensor. The sensor was stored at room temperature in an airtight box.
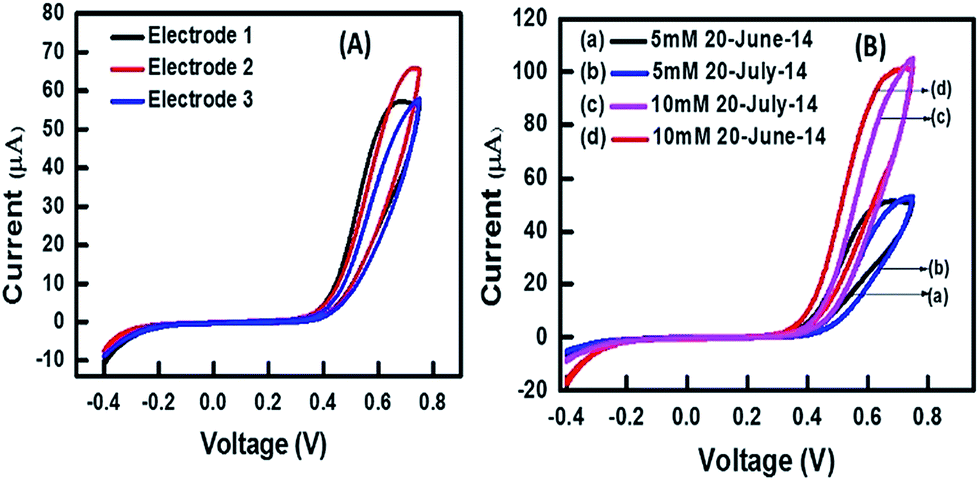 | ||
| Fig. 11 (A) CV for 3 electrodes with 5 mM H2O2. (B) CV of one electrode tested at 5 mM and 10 mM H2O2 after storage. | ||
Limit of detection (LOD) for a sensor is an important characteristic. To determine the limit of detection, the difference in current values (I − Io) were plotted for the sensor (blue line) as well as control (red line) electrode. The standard deviation for each data point was computed by using current values obtained from 3 different electrodes prepared by the same procedure as described in the experimental section. The standard deviation and respective mean values were plotted and fitted by a linear curve for both sensor as well as control. The correlation coefficients of the fitted line for sensor and control were 0.9955 and 0.9976 respectively. The limit of detection was found to be 4.22 × 10−7 M (∼0.0143 ppm) (based on signal/noise = 3).48–51 Also the standard deviations of control and sensor at the point closest to limit of detection (0.4 μM) were well separated, supporting this LOD value (Fig. 12).
 | ||
| Fig. 12 Limit of detection of the sensor (Ru@SBA15-SH deposited on Au SPE). | ||
4. Conclusions
Here, we have reported the preparation of Ru@SBA15-SH modified screen printed Au electrode for quantitative measurement of H2O2 concentration in aqueous medium. This sensor shows its capability to measure wide range of concentration of H2O2 (from 100 mM to 2 μM) reliably with high sensitivity. The detection limit of this sensor is as low as ∼4.22 × 10−7 M, which is well below the permitted standards of H2O2. To the best of our knowledge, the LOD for the sensor reported here is lowest among most of the reported electrochemical sensors for H2O2 detection. Table 1 listed the LOD values of some of the electrochemical sensors for detection of H2O2. High sensitivity, wide range of detection, good durability, reliability and high efficiency make this electrode an attractive sensor for quantitative detection of H2O2. This electrode, with some modification has also shown promising results for detection of glucose, lactose and alcohol and the results will be communicated shortly.| Sr no. | Material used | Detection method | LOD (μM) | Ref. |
|---|---|---|---|---|
| 1 | Pt/Au gold disk | CV/amperometry | 4.00 | 30 |
| 2 | Ru(bpy)32+–RuO2·xH2O | Fluorescence-optical sensor | 100 | 45 |
| 3 | Pt–Ru bimetallic nanoparticle | Amperometry | 10 | 46 |
| 4 | CPE–RuNP | Amperometry | 3.78 × 106 | 51 |
| 5 | Ag nanoparticle | Amperometry | 0.6 | 58 |
| 6 | Ag–DNA hybrid NPs | Amperometry/CV | 0.6 | 59 |
| 7 | Au NP–TiO2 nanotube | CV/chronoamperometry | 2.00 | 60 |
| 8 | HRP–graphene | Electrochemistry | 1.17 | 61 |
| 9 | HRP–Au nanoparticle | Electrochemistry | 5.90 | 62 |
| 10 | TiO2 nanotube array | Amperometry | 1.2 | 63 |
| 11 | Co3O4 nanowires | Electrochemistry | 1.4 | 64 |
| 12 | Ru@SBA15-SH on Au SPE | CV | 0.4 | This sensor |
Acknowledgements
Dr N. N. Ghosh and Prof. P. A. Millner gratefully acknowledge financial support from DST-UKIERI. The authors would like to thank Mr Areef Sardar, NIO, Goa for providing EDS measurements.Notes and references
- C. G. Tsiafoulis, P. N. Trikalitis and M. I. Prodromidis, Electrochem. Commun., 2005, 7, 1398 CrossRef CAS PubMed.
- V. Biju, Chem. Soc. Rev., 2014, 43, 737 RSC.
- C. Laloi, K. Apel, A. Danon and C. Opin, Plant Biol., 2004, 7, 323 CAS.
- W. Chen, S. Cai, Q. Ren, W. Wen and Y. Zhao, Analyst, 2010, 137, 49 RSC.
- Y. Fujita, K. Wakabayashi, M. Nagao and T. Sugimura, Mutat. Res. Lett., 1985, 144, 227 CrossRef CAS.
- L. Vitetta and A. W. Linnane, Inflammopharmacology, 2014, 22, 69 CrossRef CAS PubMed.
- M. Giorgio, M. Trinei, E. Migliaccio and P. G. Pelicci, Nat. Rev. Mol. Cell Biol., 2007, 8, 722 CrossRef CAS PubMed.
- A. Mills, C. Tommons, R. T. Bailey, M. C. Tedford and P. J. Crilly, Analyst, 2007, 132, 566 RSC.
- W. H. McCurdy Jr and H. F. Bell, Talanta, 1966, 13, 925 CrossRef CAS.
- K. Zhang, L. Mao and R. Cai, Talanta, 2000, 51, 179 CrossRef CAS.
- R. F. P. Nogueira, M. C. Oliveira and W. C. Paterlini, Talanta, 2005, 66, 86 CrossRef CAS PubMed.
- M. Hoshino, S. Kamino, M. Doi, S. Takada, S. Mitani, R. Yanagihara, M. Asano, T. Yamaguchi and Y. Fujita, Spectrochim. Acta, Part A, 2014, 117, 814 CrossRef CAS PubMed.
- Q. Chang, K. Deng, L. Zhu, G. Jiang, C. Yu and H. Tang, Microchim. Acta, 2009, 165, 299 CrossRef CAS.
- Y. Yang, Q. Li, X. Yu, X. Chen and Y. Wang, Food Control, 2014, 39, 198 CrossRef CAS PubMed.
- M. Schäferling, D. B. M. Grögel and S. Schreml, Microchim. Acta, 2011, 174, 1 CrossRef.
- S. G. Rhee, T. Chang, W. Jeong and D. Kang, Mol. Cell, 2010, 29, 539 CrossRef CAS PubMed.
- X. Chen, D. Li, H. Yang, Q. Zhu, H. Zheng and J. Xu, Anal. Chim. Acta, 2001, 434, 51 CrossRef CAS.
- T. Huang, M. E. Garceau and P. Gao, J. Pharm. Biomed. Anal., 2003, 31, 1203 CrossRef CAS.
- S. Hanaoka, J. Lin and M. Yamada, Anal. Chim. Acta, 2001, 426, 57 CrossRef CAS.
- A. Tahirović, A. Čopra, E. Omanović-Mikličanin and K. Kalcher, Talanta, 2007, 72, 1378 CrossRef PubMed.
- L. Zhang, Y. Chen, Z. Zhang and C. Lu, Sens. Actuators, B, 2014, 193, 752 CrossRef CAS PubMed.
- X. Hu, H. Han, L. Hua and Z. Sheng, Biosens. Bioelectron., 2010, 25, 1843 CrossRef CAS PubMed.
- M. Xu, W. Qi, L. Zhang, J. Lai, A. Rehman, S. Majeed and G. Xu, Microchim. Acta, 2014, 181, 737 CrossRef CAS.
- A. Chen and S. Chatterjee, Chem. Soc. Rev., 2013, 42, 5425 RSC.
- M. Holzinger, A. L. Goff and S. Cosnier, Front. Chem., 2014, 2, 2296 Search PubMed.
- J. Wang, Analyst, 2005, 4, 421 RSC.
- X. Cui, S. Wu, Y. Li and G. Wan, Microchim. Acta, 2015, 182, 265 CrossRef CAS.
- K. Cui, Y. Song, Y. Yao, Z. Huang and L. Wang, Electrochem. Commun., 2008, 10, 663 CrossRef CAS PubMed.
- N. Butwong, L. Zhou, W. Ng-eontae, R. Burakham, E. Moore, S. Srijaranai, J. H. T. Luong and J. D. Glennon, J. Electroanal. Chem., 2014, 717, 41 CrossRef PubMed.
- A. A. Karyakin, E. E. Karyakina and L. Gorton, Anal. Chem., 2000, 72, 1720 CrossRef CAS.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813 CrossRef CAS.
- N. A. Hirst, L. D. Hazelwood, D. G. Jayne and P. A. Millner, Sens. Actuators, B, 2013, 186, 674 CrossRef CAS PubMed.
- E. Nossola and A. J. G. Zarbin, J. Mater. Chem., 2012, 22, 1824 RSC.
- I. L. Mattos, L. Gorton and T. Ruzgas, Biosens. Bioelectron., 2003, 18, 193 CrossRef.
- U. Scharf and E. W. Grabner, Electrochim. Acta, 1996, 41, 233 CrossRef CAS.
- Y. Zhang and G. S. Wilson, J. Electroanal. Chem., 1993, 345, 253 CrossRef CAS.
- T. Tangkuaram, C. Ponchio, T. Kangkasomboon, P. Katikawong and W. Veerasai, Biosens. Bioelectron., 2007, 22, 2071 CrossRef CAS PubMed.
- C. Gao, Z. Gao, J. Liu and X. Huang, Nanoscale, 2012, 4, 1948 RSC.
- Y. Xiao, H. Ju and H. Chen, Anal. Chim. Acta, 1999, 391, 73 CrossRef CAS.
- X. Xu, S. Liu and H. Ju, Sensors, 2003, 3, 350 CrossRef CAS PubMed.
- S. Ledru, N. Ruillé and M. Boujtita, Biosens. Bioelectron., 2006, 21, 1591 CrossRef CAS PubMed.
- M. M. T. Khan, A. Hussain, G. Ramachandraiah and M. A. Moiz, Inorg. Chem., 1986, 25, 3023 CrossRef CAS.
- P. Li, H. Liu, J. Yang, D. Sun, Y. Chen, Y. Zhou, C. Caia and T. Lua, J. Mater. Chem. B, 2014, 2, 102 RSC.
- C. A. Marquette and L. J. Blum, Anal. Bioanal. Chem., 2008, 390, 155 CrossRef CAS PubMed.
- M. F. Dousikou, M. A. Koupparis and C. E. Efstathiou, Phytochem. Anal., 2006, 17, 255 CrossRef CAS PubMed.
- M. Janyasupab, Y. Zhang, P. Lin, B. Bartling, J. Xu and C. Liu, J. Nanotechnol., 2011, 506862, DOI:10.1155/2011/506862.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548 CrossRef CAS.
- T. Suominen, P. Uutela, R. A. Ketola, J. Bergquist, L. Hillered, M. Fine, H. Zhang, A. Laakso and R. Kostiainen, PLoS One, 2013, 8, e68007 CAS.
- A. Shrivastava and V. B. Gupta, Chron. Young Sci., 2011, 2, 21 CrossRef.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712 CrossRef.
- V. Januzaj, V. Mula, G. L. Turdean and L. M. Muresan, Acta Chim. Slov., 2015, 62, 1 Search PubMed.
- H. Xiong, Y. Zhang, S. Wang, K. Liew and J. Li, J. Phys. Chem. C, 2008, 112, 9706 CAS.
- H. Li, R. Wang, Q. Hong, L. Chen, Z. Zhong, Y. Koltypin, J. Calderon-Moreno and A. Gedanken, Langmuir, 2004, 20, 8352 CrossRef CAS PubMed.
- K. V. R. Chary and C. S. Srikanth, Catal. Lett., 2009, 128, 164 CrossRef CAS PubMed.
- H. He, X. Peng, M. Huang, G. Chang, X. Zhang and S. Wang, Analyst, 2014, 139, 5482 RSC.
- S. Rojas, E. Quartapelle-Procopio, F. J. Carmona, M. A. Romero, J. A. R. Navarro and E. Barea, J. Mater. Chem. B, 2014, 2, 2473 RSC.
- H. Li, F. Liu, S. Sun, J. Wang, Z. Li, D. Mu, B. Qiaoa and X. Peng, J. Mater. Chem. B, 2013, 1, 4146 RSC.
- Z. H. He, Y. H. Song, L. Wang, L. L. Wan, H. Z. Zhu and S. Gao, Asian J. Chem., 2012, 24, 3837 CAS.
- S. Wu, H. Zhao, H. Ju, C. Shi and J. Zhao, Electrochem. Commun., 2006, 258, 2788 Search PubMed.
- A. K. M. Kafi, G. Wu and A. Chen, Biosens. Bioelectron., 2008, 24, 566 CrossRef CAS PubMed.
- Y. Wang, X. Ma, Y. Wen, Y. Xing, Z. Zhang and H. Yang, Biosens. Bioelectron., 2010, 25, 2442 CrossRef CAS PubMed.
- H. Zhou, Q. A. Zhang, Y. Qiao, L. Zhang, S. Y. Wu, J. W. Xu and X. M. Song, Electroanalysis, 2011, 23, 900 CrossRef PubMed.
- S. Liu and A. Chen, Langmuir, 2005, 21, 8955 Search PubMed.
- M. Liu, S. He and W. Chen, Nanoscale, 2014, 6, 11769 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Wide angle XRD spectrum of Ru@SBA15 and Ru@SBA15-SH, EDS spectra of Ru@SBA15, and cyclic voltammogram for High Potential range Scan (HPS). See DOI: 10.1039/c5ra02712h |
| This journal is © The Royal Society of Chemistry 2015 |