
Development of environmentally sensitive fluorescent and dual emissive deoxyuridine analogues†
N. P. F. Barthesa,
J. Karpenko‡
a,
D. Dziuba§
a,
M. Spadaforaa,
J. Auffreta,
A. P. Demchenkob,
Y. Mélyc,
R. Benhidaa,
B. Y. Michel*a and
A. Burger*a
aInstitut de Chimie de Nice, UMR 7272, Université de Nice Sophia Antipolis, CNRS, Parc Valrose, 06108 Nice Cedex 2, France. E-mail: benoit.michel@unice.fr; burger@unice.fr
bA.V. Palladin Institute of Biochemistry, 9 Leontovicha Street, Kiev 01030, Ukraine
cLaboratoire de Biophotonique et Pharmacologie, UMR 7213, Faculté de Pharmacie, Université de Strasbourg, CNRS, 74 Route du Rhin, 67401 Illkirch, France
First published on 1st April 2015
Abstract
Ratiometric and environment-sensitive fluorescent dyes present attractive advantages for sensing interactions in DNA research. Here, we report the rational design, synthesis, and photophysical characterization of 2-thienyl-, 2-furyl- and 2-phenyl-3-hydroxychromones bonded to the C-5 position of deoxyuridine. Since these two-color nucleosides were designed for incorporation into ODNs, we also investigated the sensitivity of the ratiometric response to hydration by using acetonitrile/water mixtures and neat solvents. The synthesized 2-thienyl and 2-furyl conjugates were found to exhibit more red-shifted absorption (by 31–36 nm) and emission (by 77–81 nm of the N* band), two-fold increased molar absorption coefficients, and dramatically enhanced (by 3–4.5 times) fluorescence quantum yields. Demonstrating a manifold increase in brightness, they preserve the ability of exquisite ratiometric responses to solvent polarity and hydration. This makes the new fluorescent nucleoside analogues highly relevant for subsequent labeling of the major groove in nucleic acids and sensing their interactions.
Introduction
Fluorescence spectroscopy is a highly versatile tool widely applied in biomolecular research.1 Unfortunately, the very low intrinsic fluorescence of DNA allows only a very limited number of applications by this technique.2 In contrast, the introduction of fluorescent probes provides broad access to a variety of applications that include probing DNA hybridization,3 typing single-nucleotide polymorphism (SNP),4 and monitoring the dynamics of DNA/protein complexes,5,6 to name a few. One major approach to design a DNA-fluorescent sensor is to incorporate into DNA, a fluorescent nucleoside analogue as a fluorescent signal transducer.7 Fluorophores that exhibit extreme sensitivity to environmental changes and interactions are highly desirable to provide site-specific responses in sensing.A large number of analogues of fluorescent nucleosides, such as the widely used 2-amino-purine label (2-AP), responds to environmental changes by a variation in the intensity of fluorescence.8 However, low sensitivity to the surrounding environment and low quantum yields in DNA duplexes limit the use of such intensiometric probes. These limitations encourage investigating the synthesis of advanced emissive nucleosides based on new mechanisms of response. Ratiometric sensing, also referred to as λ-ratiometric,9 is one such approach, and is based on recording a ratio of the intensities at two or more wavelengths. Compared to simple intensity sensing, ratiometric sensing is advantageous since it compensates for instrumental factors such as fluctuations in the intensity of the light source, and because the resulting intrinsically calibrated analytical response does not depend on the concentration of the applied dye.10 To this point, two-channel probing was mainly based on double-labeling of nucleic acids as for the construction of Förster resonance energy transfer (FRET) and excimer pairs.3e,f,4j,5c Difficulties and cost of synthesis obviously represent limitations for this approach. Alternatively, λ-ratiometric sensing can be performed with a single fluorophore9 using environment-sensitive probes. One class of these probes comprises single-band solvatochromic dyes, which respond by a spectral shift due to an excited-state intramolecular charge transfer (ICT). However, these single λ-ratiometric emitters remain faintly explored in the field of DNA sensing11 mainly due to the strong variations of the intensity of fluorescence that accompany the shifts. Dual emissive fluorophores emerge as a second class. These dyes exhibit two emission bands as a result of twisted excited-state intramolecular charge transfer (TICT)12 or excited-state intramolecular proton transfer (ESIPT).13 Dual emissive dyes are very attractive because they offer facile and straightforward quantification through the ratio of their two bands. Nevertheless, they are still poorly exploited because of the very limited number of dyes that display such properties and the difficulties in their syntheses.
Among fluorophores with dual emission, 3-hydroxychromones (3HCs) appear particularly promising in terms of photophysics and applications as biosensor units. Due to the ESIPT reaction, 3HCs exhibit two excited states, the normal form (N*) and the tautomer form (T*), that provide two well separated emission bands (Fig. 1).14 3HC fluorophores offer exceptional solvatochromism,15 since both an increase in the hydrogen bond donor strength and the dielectric constant of solvents inhibit the ESIPT reaction and thus, decrease the relative intensity of the T* band.16 Therefore, changes in the polarity and the hydration of the microenvironment of 3HCs can be monitored by measuring the ratio of the intensities of the two emission bands (IN*/IT*).15 This unique behaviour of 3HCs was the cornerstone for the design of environment-sensitive fluorescent probes for a large range of applications. These include the characterization of supercritical fluids,17 micelles,18 phospholipids bilayers,19 plasma membranes of living cells,20 and biomolecular interactions.21,22 Their applications as fluorescent nucleoside analogues to label oligonucleotides (ODNs) has only been recently explored.23,24
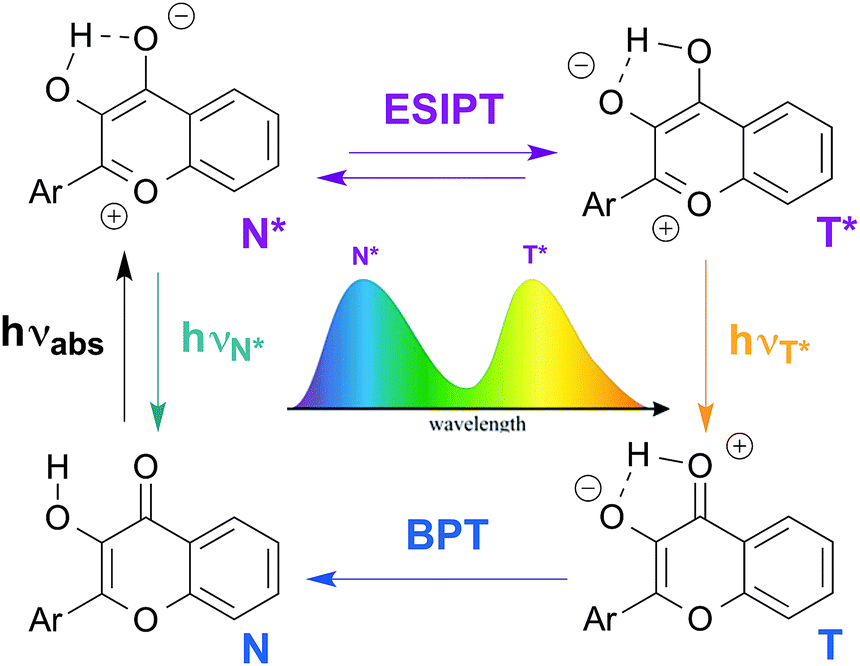 | ||
| Fig. 1 Excited state intramolecular proton transfer (ESIPT) reaction of 3-hydroxychromones: genesis of the dual emission. BPT: back proton transfer. | ||
The fluorescent C-nucleoside 1 using 2-thienyl-3-hydroxychromone moiety was first designed as a substitute for natural bases for internal labeling of ODNs and sensing (Fig. 2).25,26 As a next step, external labeling of DNA major groove was addressed by compiling 2-thienyl-3-hydroxychromone and uracil at the 5-position via an ethynyl bond.26 The synthesized conjugate demonstrated that the nucleobase and 3HC fragments were coupled into an electronic conjugated system, which attained strong ICT character upon excitation.
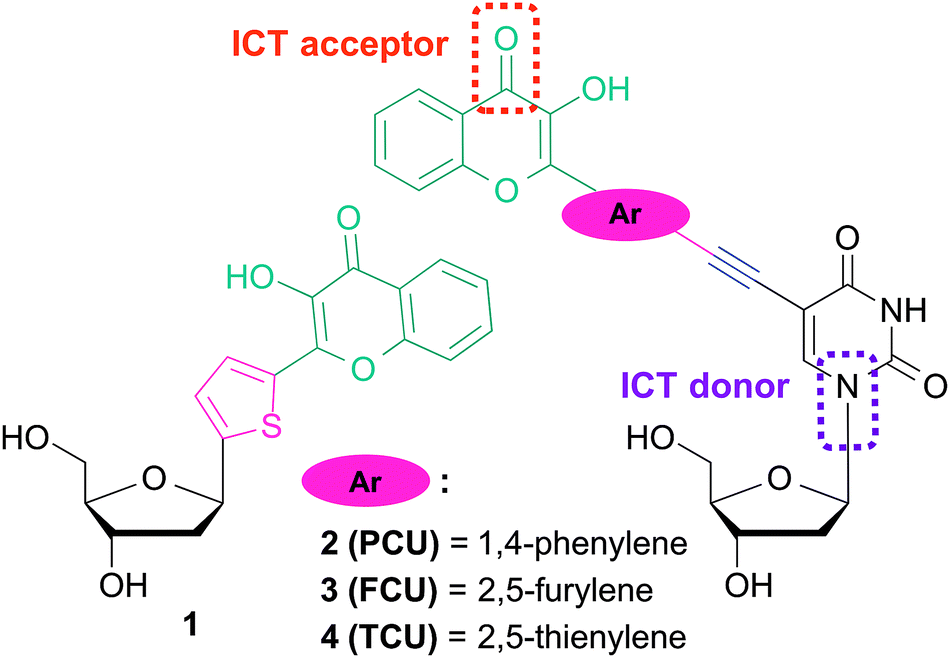 | ||
| Fig. 2 Fluorescent nucleoside analogues incorporating 3-hydroxychromone fluorophore as a nucleobase surrogate 123,24 and natural base modifier 2–4.25 | ||
The ICT states are crucial in achieving the solvent-dependent two-band emission of 3HC dyes. The substitutions in the position 2 of the chromone demonstrates the strongest modulating power27a that allows reaching a dynamic equilibrium between the populations of ICT and ESIPT states, from which the dual emission originates.27b With the aim to label DNA and to tune the spectroscopic properties of the conjugates for sensing, reliable synthesis of 2′-deoxyuridine conjugates bearing aryl groups of different natures connected to the chromone was developed (Fig. 2). Herein, we report the synthesis and the photophysics of 2′-deoxyuridine coupled to 2-phenyl-, 2-furyl- and 2-thienyl-3-hydroxychromone referred as PCU, FCU and TCU, respectively (2–4).
Results and discussion
The 3HC deoxyuridines PCU, FCU, and TCU were furnished by a convergent synthetic strategy based on Sonogashira couplings and Algar–Flyn–Oyamada reaction (Scheme 1). Sonogashira reaction28 is widely applied to couple aromatic scaffolds, including dyes such as pyrenes,12b,29 fluorenes,30 naphthalenes and anthracenes,31 to deoxyuridine. This palladium-catalyzed cross-coupling proceeds usually by assembling a C-5 iodinated uracil moiety with an aromatic alkyne.32 Therefore, the ethynyl moiety is generally introduced to the aromatic molecule through a cross-coupling with TMS-acetylene, after which the alkyne is deprotected from its silyl group and directly coupled to the heterocyclic 5-iodo-2′-deoxyuridine derivative.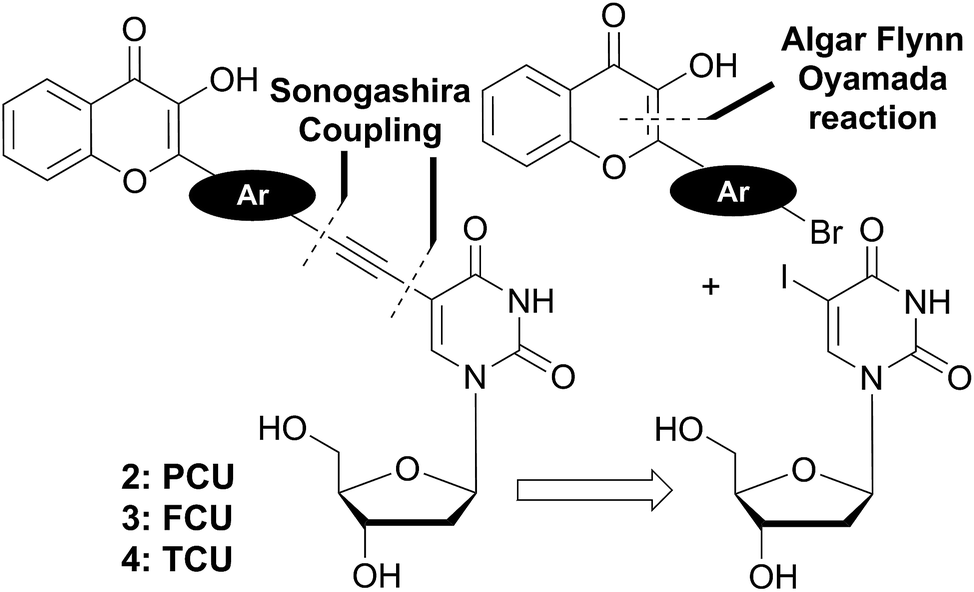 | ||
| Scheme 1 Retrosynthetic analysis of the targeted two-color emission nucleosides 2–4. | ||
We applied this approach for the preparation of the target compounds. However, we faced difficulties in the elaboration of ethynylchromones (Scheme S1†), and found that the Sonogashira coupling between our tested ethynylchromone and 5-iodo-2′-deoxyuridine was sluggish and low yielding. This might be attributed to a competition between homo- and cross-couplings. Indeed, it has been known that homo-coupling is favored in the presence of electron-withdrawing groups on aryl alkynes.28 So the 3HC substitution brings electron deficiency to the terminal alkyne, consequently increasing the competition of the homo- and cross-couplings to access the catalyst. To overcome these difficulties, we adopted an alternative approach based on reacting the electron-rich ethynyl-deoxyuridine derivative with the electron-poor bromo-arylchromones. The new suggested strategy established a more reliable synthetic access to the target compounds.
The preparation of the 3HC key intermediates 9–15 required for the subsequent coupling with the 5-ethynyldeoxyuridine derivative is described in Scheme 2. The bromochromones 9–11 were obtained using the fast and versatile Algar–Flynn–Oyamada reaction.33 Thus, starting from 2-hydroxy-acetophenone 5, an aldol condensation/dehydratation process with aldehydes 6–8 in the presence of sodium hydroxide in ethanol followed by an oxidative cyclization with aqueous hydrogen peroxide provided the chromones 9–11 (48–58%).
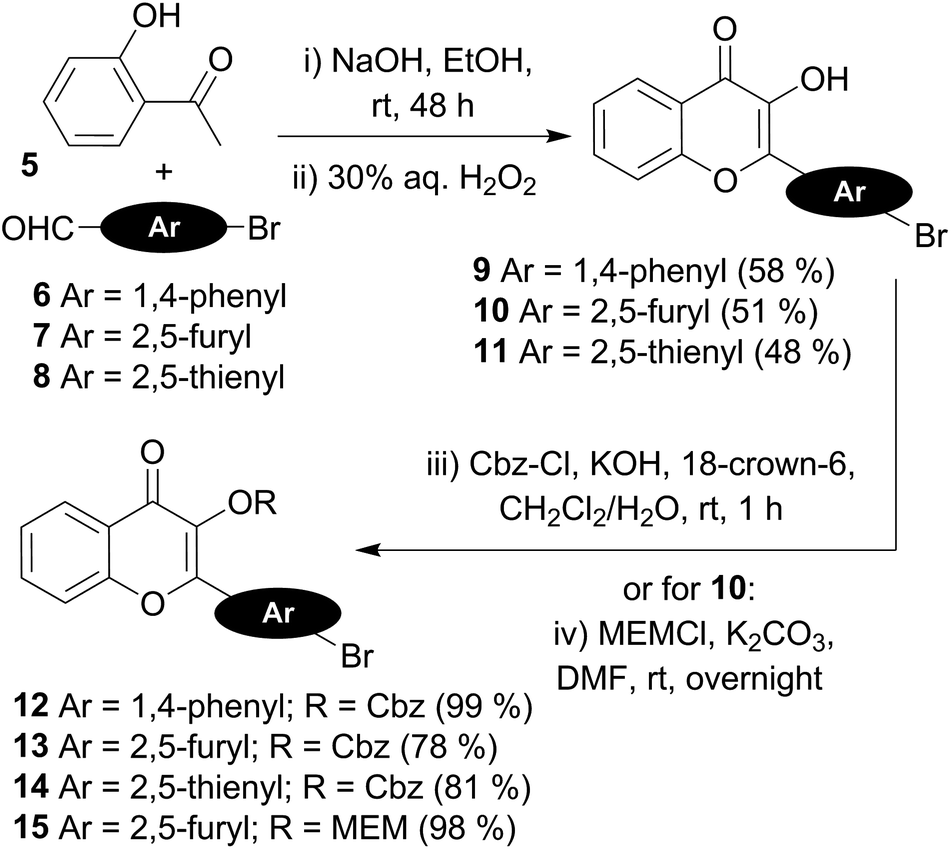 | ||
| Scheme 2 Synthetic preparation of the fluorescent chromone-coupling partners. | ||
The synthesis of the chromone–deoxyuridine conjugates required masking the reactive 3-OH group of the chromone. The benzyloxycarbonyl group (Cbz), which was successfully employed during the synthesis of 3HC-containing C-nucleosides and ODNs,24 and the more robust methoxyethoxymethyl group (MEM)25 were chosen as the protecting groups. The Cbz protection of the 3-OH group of chromones 9–11 was performed under phase-transfer catalysis conditions,34 delivering compounds 12–14 in good yields. Protection with the MEM group was obtained by treatment of the furyl chromone 10 with potassium carbonate and MEMCl in DMF, producing compound 15.
Next, we synthesized the 5-ethynyldeoxyuridine partner 18, using common procedures (Scheme 3). The protection of deoxyuridine 16 before the iodination of the nucleobase at the C-5 position with cerium(IV) ammonium nitrate turned out be more convenient and efficient.35 Sonogashira coupling of the iodo intermediate 17 with TMS-acetylene followed by treatment with tetrabutylammonium fluoride furnished the terminal alkyne 18 in satisfactory yields (64% in 2 steps).¶
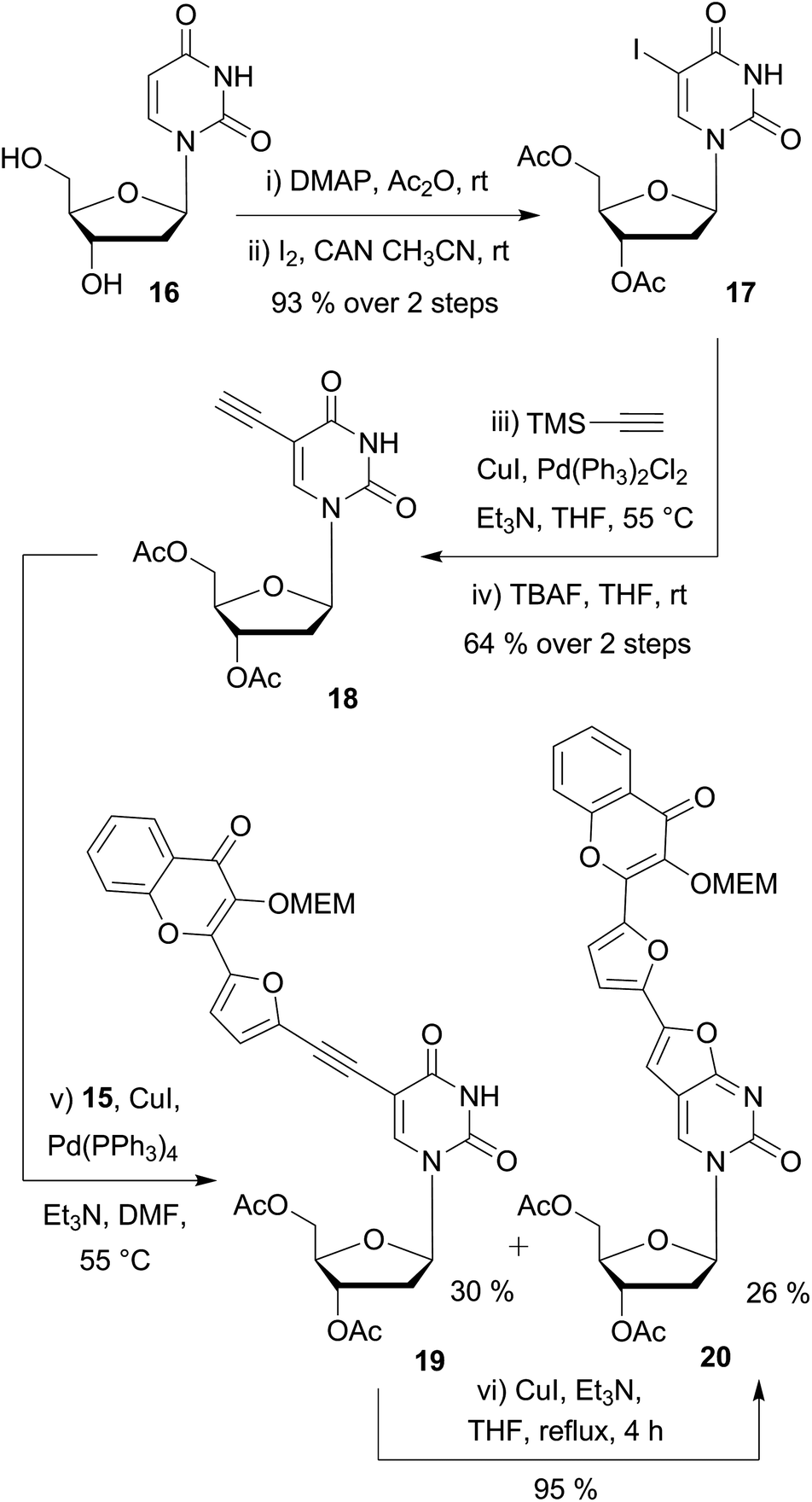 | ||
| Scheme 3 Preliminary Sonogashira couplings on substrate 18 having no N3-protecting group on the uracil moiety. | ||
Preliminary attempts of the final assembly were performed with the protected bromochromone 15 (Scheme 3). Different conditions were examined including the catalyst (Pd(PPh3)4 or PdCl2(PPh3)2), the loading (5–20 mol%), and the solvent (toluene, dioxane, THF and DMF). All screenings converged to two major products: the desired coupling derivative 19 and the 5-endo-dig cyclization side-product 20. Despite the use of DMF, which is reported to minimize the formation of the bicyclic furanopyrimidine,36 the formation of the endo product could not be avoided. The ethynyl-coupled product 19 was difficult to purify and thus, was obtained in 30% yield after silica gel and reverse phase chromatographies. NMR and MS analysis,|| and chemical means supported the proposed structure of the endo product. As reported amongst others37 by McGuigan et al.,38–40 after the Sonogashira coupling, an extended treatment with CuI/Et3N in THF or MeOH at reflux for several hours enabled us to fully convert the coupled alkynyl derivative 19 into the 5-endo cyclized side product 20 (Scheme 3).
Endo-cyclisation of 5-ethynyluridine derivatives is a well-known reaction that is favored by prolonged reaction time at elevated temperatures, high CuI loading, and the presence of electron-withdrawing groups on the coupling partner.41 Although the copper-free Sonogashira coupling was widely reported in the last decade,42 no product at all was formed when copper-free conditions were applied to the reaction of acetylenic and iodo partners as evidenced by TLC. Protection of the N3-imide with the base labile benzoyl group is known to prevent 5-endo cyclisation.43 Consequently, we selected this type of protection, using p-toluoyl rather than benzoyl, since the former presents the advantage of displaying a characteristic methyl singlet easy to detect by NMR. To set up the imide protecting group, mild conditions (p-tolyl chloride, Et3N in pyridine) were employed to provide 21 (82%) as the building block for the final assembly of the two fragments (Scheme 4). Second Sonogashira coupling was then efficiently used for that purpose. Thus, when using a standard catalytic system (PdCl2(PPh3)2, CuI, Et3N in THF), the reaction proceeded cleanly to give the desired coupled derivatives 22–24. It is noteworthy that the yields of C(sp)–C(sp2) cross coupling for the five-membered ring partners 23 and 24 (82 and 87%) are more satisfactory as compared to the one obtained with the phenyl moiety 22 (60%). The lower yield of the phenyl derivative could be attributed to the higher steric hindrance of the six membered ring and/or to the lower reactivity of the more electronically poor bromophenyl moiety with the electron withdrawing chromone. A final deprotection of the esters, carbonate and amide groups via a simple treatment with aqueous ammonia provided the three targeted conjugated fluorescent compounds 2 (PCU), 3 (FCU) and 4 (TCU), which were used for photophysical characterizations.
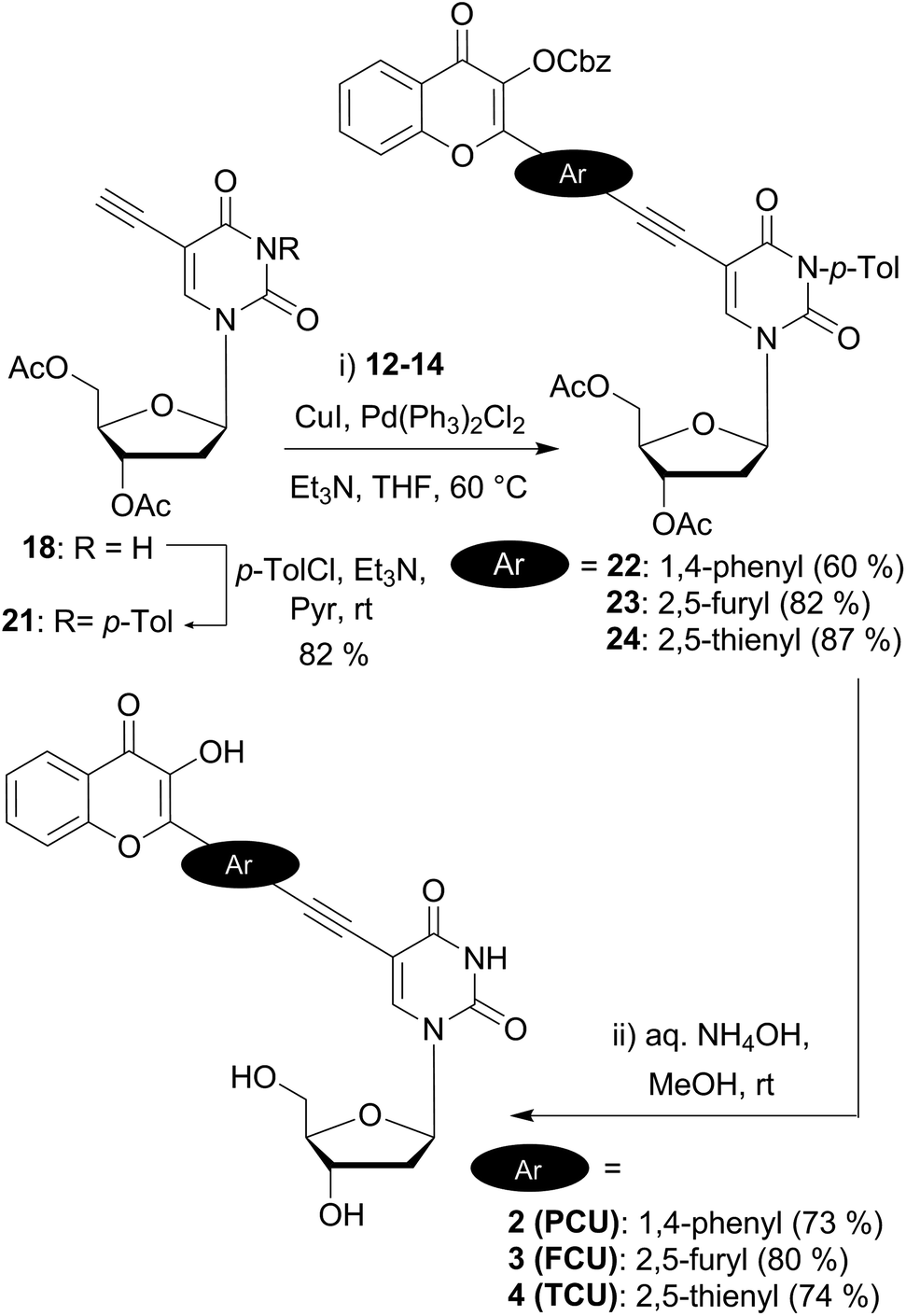 | ||
| Scheme 4 Synthesis of dual emissive fluorescent nucleosides 2, 3 and 4 bearing a 3HC scaffold. | ||
Photophysical studies
Results
UV/Vis absorption and fluorescence properties of the three chromone-nucleoside analogues PCU, FCU, and TCU were investigated in methanol and compared with those of the corresponding parent 2-phenyl, 2-furyl and 2-thienyl-3-hydroxychromones, PC, FC, and TC respectively (Table 1, Fig. S1†).| Dye | λ abs a | Δλabsb | ε c | ε π/εχd | λ N* e | ΔλN*f | Φ g | Φ π/Φχh |
|---|---|---|---|---|---|---|---|---|
| a Position of the absorption maximum in nm. b Difference of absorption maxima between the conjugate and the parent 3HC in nm. c Molar absorption coefficient in M−1 cm−1. d Ratio of molar absorption coefficients between the conjugate (επ) and the parent (εχ) 3HC. e Position of the normal N* band maximum in nm. f Difference of emission maxima between the conjugate and the parent 3HC in nm. g Quantum yields determined using 3-hydroxyflavone in toluene (Φ = 0.29),47 4′-(dimethylamino)-3-hydroxyflavone in ethanol (Φ = 0.27)49 or quinine sulfate in 0.1 M H2SO4 (Φ = 0.54)50 as standard references. h Ratio of quantum yields between the conjugate (Φπ) and the parent (Φχ) 3HC. | ||||||||
| PC 46,47 | 343 | — | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 300 300 |
— | 403 | — | 0.03 | — |
| PCU | 368 | 25 | 27000 |
2 | 500 | 97 | 0.30 | 10 |
| FC 46,48 | 356 | — | 16400 |
— | 416 | — | 0.08 | — |
| FCU | 387 | 31 | 37000 |
2.2 | 497 | 81 | 0.24 | 3 |
| TC 26 | 359 | — | 23500 |
— | 424 | — | 0.07 | — |
| TCU | 395 | 36 | 39000 |
1.7 | 501 | 77 | 0.32 | 4.5 |
These studies brought interesting results. In point of fact, the positions of the light absorption bands of the new derivatives were located in the same sequence as that of the parent 3HCs but demonstrated red-shifts by 25–36 nm, which could be explained by an electronic conjugation with uracil moiety (Fig. 3). Such conjugation should increase dramatically the light absorption cross-section leading to increase correspondingly the molar absorption coefficients. PCU, FCU, and TCU absorbed light about twice stronger, with molar absorption coefficients of about 27000, 37000 and 39000 M−1 cm−1. It must be noted that the shift of the absorption maxima to the red, and the increase of the molar absorption coefficients proceed in the following sequence: PCU < FCU < TCU. The more red-shifted absorption and emission of the thienyl and furyl derivatives as well as the increased absorption coefficients are in agreement with the higher electronic density of substituents in position 2 of the chromone ring, respectively. In our case, this may result in more efficient electronic coupling between the chromone and uracil electronic systems. The differences with the phenyl substituent can be accounted for the fact that the five-membered rings are more electron rich and allow to adopt a more planar conformation with respect to chromone for better coupling.44 Thus, the novel conjugates display substantial shifts to the red of fluorescence spectra, for instance in methanol, the shifts of N* and T* bands are ca. 80 and 25 nm, respectively (Table 1, Fig. 3). The fluorescence quantum yields were also increased effectively, so that the brightness (defined commonly as the product of molar absorbance and quantum yield) could be increased roughly by one order of magnitude.
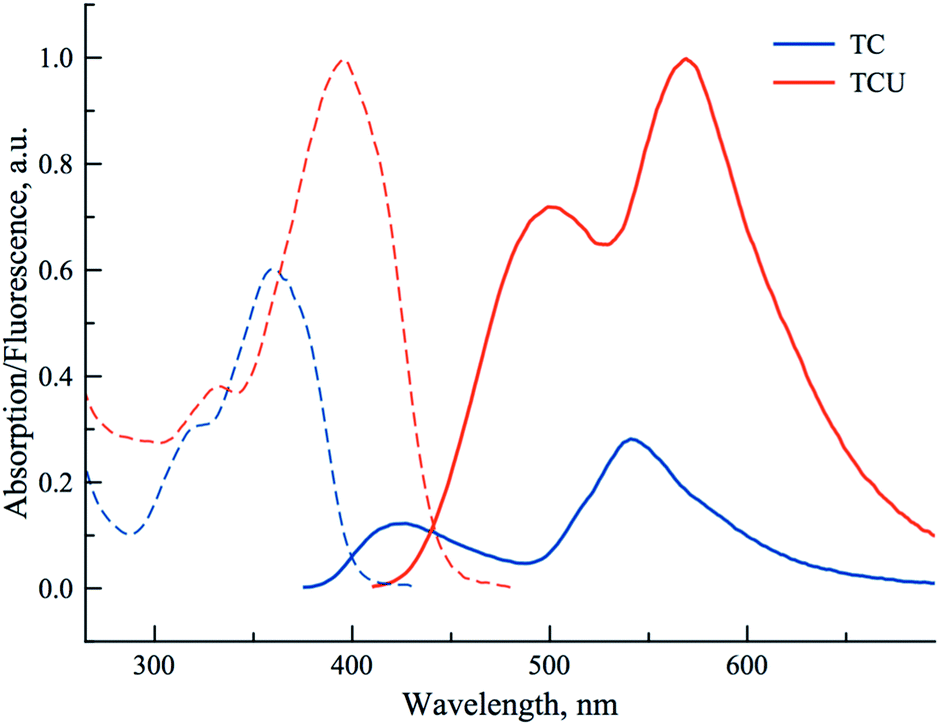 | ||
| Fig. 3 Absorption and fluorescence emission spectra of TC (blue), and TCU (red) in methanol (excitation wavelength corresponds to the absorbance maximum of each compound). Absorptions and emissions are proportional to their absorption coefficients and quantum yields, respectively. | ||
On the next step, the properties of PCU, FCU and TCU conjugates were investigated and compared in a set of solvents presenting a large range of polarities (Table 2). For these studies, five aprotic solvents (acetonitrile, acetone, chloroform, ethyl acetate, toluene) and four protic solvents (hexafluoropropan-2-ol (HFIP), H2O, MeOH, EtOH) were selected.
| Solvent | E T(30)a | λ N* b | λ T* c | I N*/IT*d | Φ e | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PCU | FCU | TCU | PCU | FCU | TCU | PCU | FCU | TCU | PCU | FCU | TCU | ||
| a Reichardt's empirical solvent polarity index.45 b Position of the N* band maximum in nm. c Position of the T* band maximum in nm. d Ratio of the two intensities. e Quantum yields determined using 3-hydroxyflavone in toluene (Φ = 0.29),47 4′-(dimethylamino)-3-hydroxyflavone in ethanol (Φ = 0.27)49 or quinine sulfate in 0.1 M H2SO4 (Φ = 0.54).50 | |||||||||||||
| HFIP | 65.3 | 472 | 493 | 500 | — | — | — | — | — | — | 0.28 | 0.33 | 0.40 |
| H2O | 63.1 | 480 | 499 | 502 | 525 | — | — | 0.74 | — | — | 0.05 | 0.13 | 0.06 |
| MeOH | 55.4 | 500 | 497 | 504 | 547 | 551 | 568 | 0.64 | 1.03 | 0.79 | 0.30 | 0.24 | 0.32 |
| EtOH | 51.9 | 489 | 488 | 501 | 552 | 566 | 575 | 0.33 | 0.56 | 0.44 | 0.26 | 0.22 | 0.23 |
| CH3CN | 45.6 | 445 | 466 | 474 | 549 | 569 | 580 | 0.03 | 0.20 | 0.28 | 0.33 | 0.17 | 0.19 |
| Acetone | 42.2 | 455 | 462 | 469 | 553 | 574 | 583 | 0.04 | 0.17 | 0.29 | 0.22 | 0.16 | 0.25 |
| CHCl3 | 39.1 | 426 | 454 | 459 | 543 | 564 | 572 | 0.02 | 0.15 | 0.17 | 0.40 | 0.23 | 0.19 |
| EtOAc | 38.1 | 424 | 444 | 457 | 551 | 573 | 582 | 0.03 | 0.10 | 0.20 | 0.34 | 0.22 | 0.20 |
The Reichardt's polarity parameter ET(30) that accounts for the dielectric constant of the solvent and its H-bond donor ability was employed to scale the polarity of the solvents.45 We observed that the positions of the absorption maxima were similar to the ones observed in MeOH (Table 1). Therefore, they correlate poorly with the solvent polarity displaying almost no solvatochromism, which is the evidence of an absence of charge-transfer character in their ground states.
PCU presented a dual emission in protic solvents, except HFIP. The short and long wavelength bands can be attributed to the N* and T* states. In contrast, it presented almost exclusively the T* band in all aprotic solvents (Fig. 4). In all solvents, except water and HFIP, FCU and TCU exhibited the two emission bands (Fig. 4). The positive solvatochromism of the N* band is typical for ESIPT probes with ICT character.26,27 The ICT character can be further estimated using the polarity scale of Lippert (Table S1 and Fig S5, S6†).1a,51 Plotting the Stokes shifts as a function of the orientation polarizability of the aprotic solvents demonstrated linear fits with positive slopes for both compounds. These results are compatible with an increase of the dipole moments of the normal excited states of FCU and TCU after Franck–Condon light absorption. The position of their T* band maxima appeared blue-shifted in protic solvents, as compared to aprotic ones. The blue shift can be explained by the formation of an H-bond between the 3-phenoxide oxygen in the T* state and the donor proton in the solvent.52,53
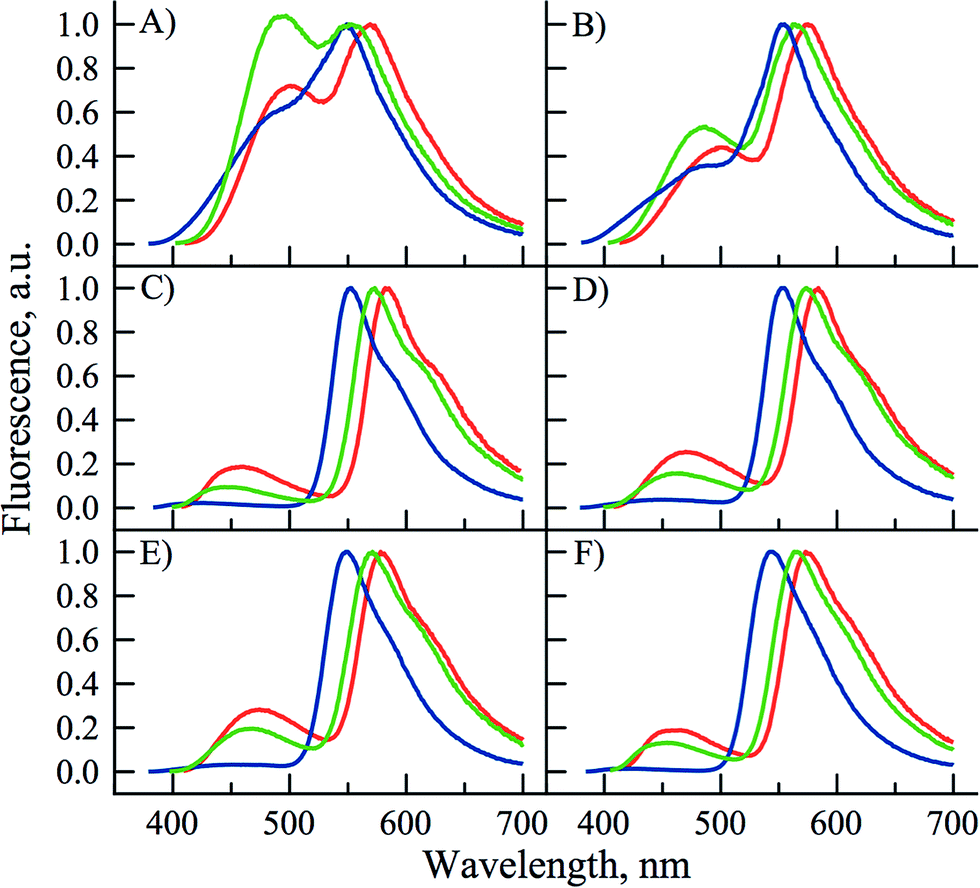 | ||
| Fig. 4 Normalized fluorescence spectra of PCU (blue), FCU (green) and TCU (red) in: (A) methanol, (B) ethanol, (C) ethyl acetate, (D) acetone, (E) acetonitrile and (F) chloroform. All the spectra are normalized by T* band (excitation wavelength corresponds to the absorbance maximum of each compound). | ||
All the three compounds displayed an increase in the IN*/IT* ratio by rising solvent polarity and H-bond donor strength (Table 2 and Fig. 4). However, FCU and TCU were much more sensitive than PCU. As a general trend, the IN*/IT* ratio of FCU and TCU gradually increased when going from the most apolar aprotic solvent to the most polar protic solvent (Table 2 and Fig. S2–S4†). It is worth indicating that the effect of H-bonding dominates the polarity effect, as that could be seen from the 3- to 5-fold larger value of the IN*/IT* ratio in methanol as compared to acetonitrile, though both solvents exhibit similar dielectric constants. Analysing the plots of Lippert further supports this interpretation. In protic solvents, the Stokes shifts were deviated to the red from the linear function indicating specific interactions between the protic solvent and the 3HC (Fig. S5 and S6†). The deviation from the linear fit in protic solvents is known for donor–acceptor solvatochromic dyes.1a The sensitivity of FCU and TCU is typical of the 3HCs exhibiting significant ICT characters, such as 2-(4-methoxy)phenyl-3HCs54 and 2-(4-dialkylamino)phenyl-3HCs.16,55
For PCU, the contribution of the N* form was marginal in aprotic solvents hinting that PCU was mainly sensitive to protic solvents that donate protons for intermolecular H-bonding with the carbonyl at position 4 of 3HC. PCU behaves like the parent 3-hydroxyflavone,56 but with a lower resolution of its dual emission.
The quantum yields of the three dyes were significantly enhanced for all the compounds in comparison with their parent analogs (Table 1). As a function of solvent, they ranged from 5 to 40%. Quantum yields were in the range of 22 to 40% in all the tested alcohols and 5 to 13% in water. However, the quantum yields in HFIP, which is more polar and acidic than water,57 were about 3 to 7 times larger than in water. Therefore, the reduced quantum yields in water were unlikely due to the H-bond strength between the donating water molecule and the accepting oxygen carbonyl of 3HC (Scheme 5). Reduction of quantum yields in water for solvatochromic dyes exhibiting strong ICT character is common. It was proposed that several water molecules act cooperatively to capture electrons from the excited-state species resulting in quenching of fluorescence.58 Though being reduced in comparison with other solvents, the quantum yields of the three compounds are still significant in water making them attractive for sensing hydrated biological media. This is a major difference as compared to many other solvatochromic dyes, which are severely quenched in water.59
 | ||
| Scheme 5 Proposed mechanism for water sensing by 3HCs adapted from previous works.52,60 | ||
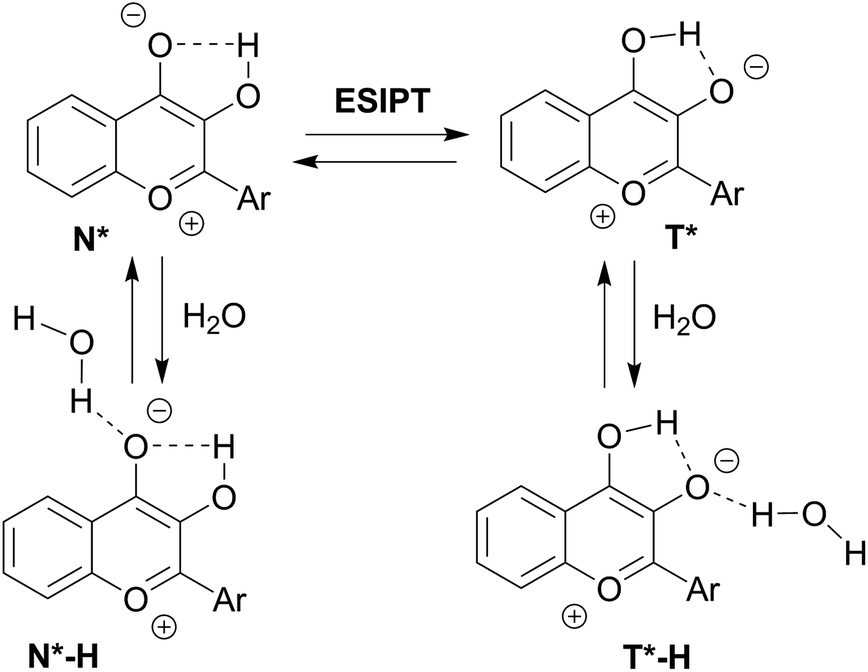
To get further insight on the sensitivity of the photophysical properties of PCU, FCU, and TCU on hydration, emission spectra were recorded in mixtures of acetonitrile and water (Fig. 5). The same set of experiments was also performed in dioxane/water mixtures in order to determine the contribution from the organic solvent to the spectral changes (Fig. S7†). A dual emission was evident in all the tested compositions for PCU and up to 70% and 90% water for FCU and TCU, respectively. In all cases, hydration increased the N*/T* ratio and shifted the T* band position to the blue, as it was observed in neat protic solvents (Table 2). The ratios of the intensities correlated linearly with the water concentration demonstrating the absence of high-affinity specific hydration (Fig. 6).
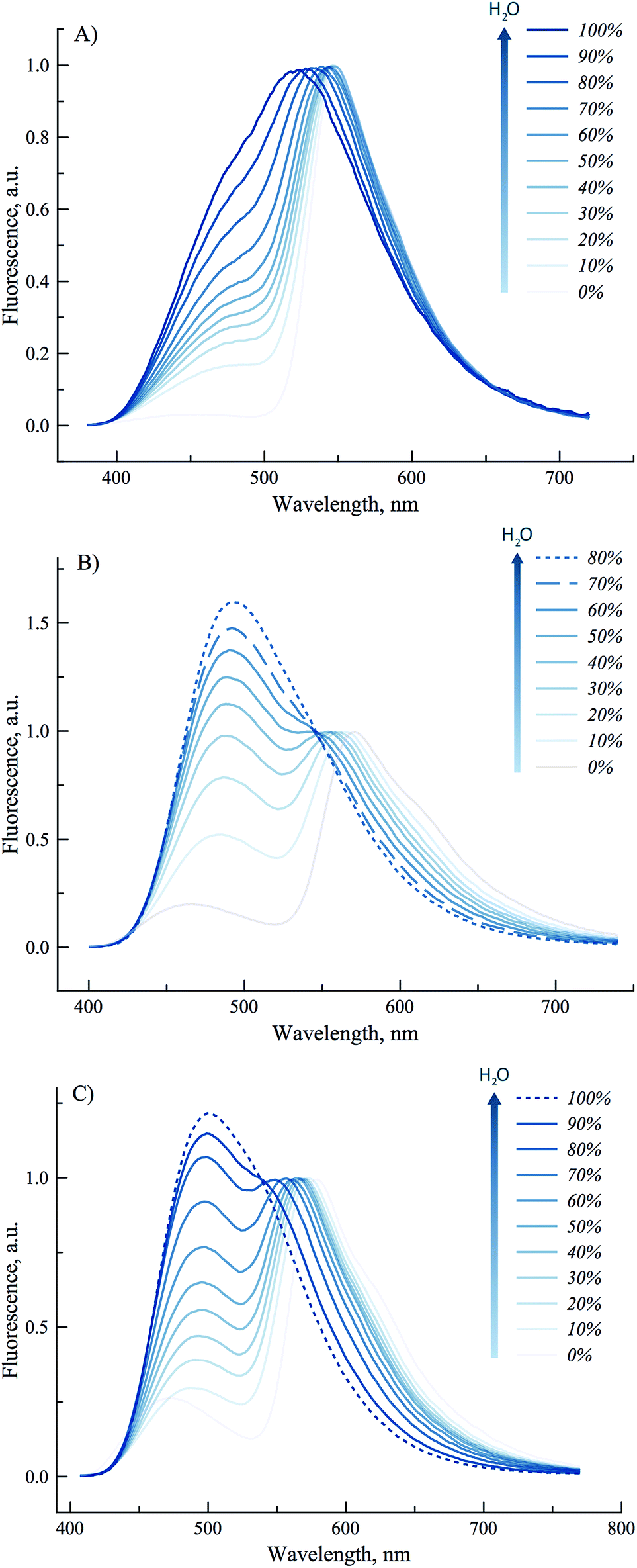 | ||
| Fig. 5 Sensitivity of the emission spectra to hydration for (A) PCU, (B) FCU and (C) TCU. Data were recorded in gradual mixtures of acetonitrile and water. Emission spectra were normalized by T* band. | ||
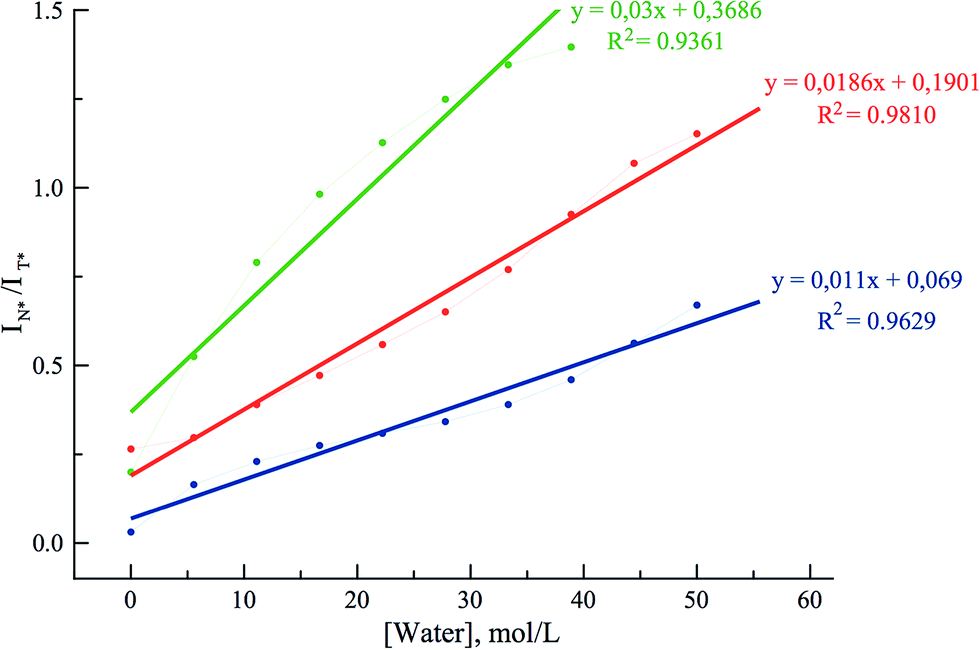 | ||
| Fig. 6 Dependence of the ratiometric response (IN*/IT*) of PCU (blue), FCU (green), and TCU (red) on the water concentration in acetonitrile. The ratios were extracted from the spectra in Fig. 4. | ||
These ratios increased gradually from 0.03, 0.2 and 0.27 in neat acetonitrile to 0.77, 1.4 and 1.15 in 100, 70 and 90% water for PCU, FCU and TCU, respectively (Fig. 5, 6 and Table S2†). To conclude, the sensitivity of the N*/T* ratio to water followed the order: FCU > TCU > PCU (slope comparison in Fig. 6). Comparable variations of the ratios of the intensities were found when dioxane was used as an organic solvent (Table S2†), confirming that the N*/T* ratio was mainly controlled by water (Fig. S8 and S9†).
The remarkable sensitivity of 3HCs to hydration correlates with previous studies carried out with derivatives of 3HCs for which a model was proposed.52,56,60 This latter involves specific H-bonding interactions of water with the electron rich oxygens of 4-carbonyl and phenoxide of 3HC (Scheme 5). As a result, the N* form exists in equilibrium with a protonated N*–H form, in which intermolecular H-bonding of the 4-carbonyl with water inhibits the ESIPT process.61 Thus, growing water concentration favors the N*–H form, and consequently, increases the N*/T* ratio. Moreover, hydration also favors H-bonding of the tautomer T* form, which results in its blue-shifted emission maximum.52
Conclusions
We developed three dual emissive nucleoside derivatives based on the assembly of a 2-aryl-3HC dye and a 2′-deoxyuridine fragment, connected through a rigid and electron-conducting ethynyl linker. The three conjugates PCU, FCU and TCU differ from each other by the C-2 aromatic ring (phenyl, furyl and thienyl) of the 3HC. Their photophysical properties were investigated in a wide range of solvents. The conjugation of furyl- and thienyl-chromones with the uracil moiety resulted in the most dramatic improvement of their spectroscopic properties.In the present work, our results show the amplification of the brightness by about one order of magnitude due to two-fold increase of the absorbance and 4 times the enhancement of the fluorescence quantum yield, whilst retaining the most remarkable property of 3HC dyes to respond to polarity and hydration of their environment by changes in their dual emission.13,16 The enhancement of the quantum yield is quite unexpected, since an increase of the conjugation in the π-electronic systems generally results in an increase of the fluorescence quenching.62 Additionally, these derivatives keep significant quantum yields in water, which is not common among polarity-sensitive dyes59,63 but was observed for a series of 3HC derivatives.46,54 This makes our new compounds highly prospective for applications as fluorescent reporters in biological media. Since water plays a key role in controlling nucleic acid structure and function through H-bonding and electrostatic interactions, the exquisite ratiometric sensitivity of FCU and TCU to hydration makes them highly relevant for subsequent major groove labeling. Moreover, the ethynyl linker is thought to locate the dye in a precise position with respect to the DNA major axis, which is of key importance for further data interpretation of biomolecular interaction studies. Incorporation of these prospective candidates into oligonucleotides, as environmentally sensitive fluorescent (ESF) probes, is currently the focus of our ongoing researches and will be reported in due time.
Acknowledgements
We thank the ANR (ANR-12-BS08-0003-02), PACA region (DNAfix-2014-02862 and 2014 07199) and the FRM (DCM20111223038) for financial support and a grant for N.P.F.B. We thank the French Government for the Master 2 grant to I.A.K. We thank Janah Shaya for critical reading of the manuscript and Dr Gilles Lemière for fruitful discussion.Notes and references
- (a) J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, New York, 3rd edn, 2006, p. 954 CrossRef; (b) B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, Wiley-VCH, Weinheim, 2nd edn, 2012, p. 592 CrossRef; (c) A. P. Demchenko, Introduction to Fluorescence Sensing, Springer, Heidelberg, 2009, p. 612 CrossRef; (d) A. P. Demchenko, Advanced Fluorescence Reporters in Chemistry and Biology III: Applications in Sensing and Imaging, Springer-Verlag, Berlin Heidelberg, 2011, p. 352 CrossRef; (e) W. T. Mason and J. I. Gallin, Fluorescent and Luminescent Probes for Biological Activity, Academic Press, 2nd edn, 1999, p. 647 Search PubMed; (f) L. Brand and M. Johnson, Methods in Enzymology: Fluorescence Spectroscopy, Academic Press, 2008, vol. 450, p. 400 Search PubMed.
- (a) M. Daniels and W. Hauswirth, Science, 1971, 171, 675–677 CrossRef CAS PubMed; (b) J. Pecourt, J. Peon and B. Kohler, J. Am. Chem. Soc., 2000, 122, 9348–9349 CrossRef CAS; (c) E. Nir, K. Kleinermanns, A. L. Grace and M. S. de Vries, J. Phys. Chem. A, 2001, 105, 5106–5110 CrossRef CAS.
- (a) J. Guo, J. Ju and N. J. Turro, Anal. Bioanal. Chem., 2012, 402, 3115–3125 CrossRef CAS PubMed; (b) A. Nadler, J. Strohmeier and U. Diederichsen, Angew. Chem., Int. Ed., 2011, 50, 5392–5396 CrossRef CAS PubMed; (c) M. E. Hawkins and F. M. Balis, Nucleic Acids Res., 2004, 32, e62–e62 CrossRef PubMed; (d) S. P. Sau and P. J. Hrdlicka, J. Org. Chem., 2011, 77, 5–16 CrossRef PubMed; (e) C. Holzhauser and H.-A. Wagenknecht, Angew. Chem., Int. Ed., 2011, 50, 7268–7272 CrossRef CAS PubMed; (f) E. Socher, L. Bethge, A. Knoll, N. Jungnick, A. Herrmann and O. Seitz, Angew. Chem., Int. Ed., 2008, 47, 9555–9559 CrossRef CAS PubMed.
- (a) S. Kim and A. Misra, Annu. Rev. Biomed. Eng., 2007, 9, 289–320 CrossRef CAS PubMed; (b) K. Tainaka, K. Tanaka, S. Ikeda, K.-I. Nishiza, T. Unzai, Y. Fujiwara, A. I. Saito and A. Okamoto, J. Am. Chem. Soc., 2007, 129, 4776–4784 CrossRef CAS PubMed; (c) Z.-Y. Zhao, M. San, J.-L. H. A. Duprey, J. R. Arrand, J. S. Vyle and J. H. R. Tucker, Bioorg. Med. Chem. Lett., 2012, 22, 129–132 CrossRef CAS PubMed; (d) J.-L. H. A. Duprey, Z.-Y. Zhao, D. M. Bassani, J. Manchester, J. S. Vyle and J. H. R. Tucker, Chem. Commun., 2011, 47, 6629–6631 RSC; (e) K. Furukawa, M. Hattori, T. Ohki, Y. Kitamura, Y. Kitade and Y. Ueno, Bioorg. Med. Chem., 2012, 20, 16–24 CrossRef CAS PubMed; (f) H. Zhang, M. Wang, Q. Gao, H. Qi and C. Zhang, Talanta, 2011, 84, 771–776 CrossRef CAS PubMed; (g) M. Hattori, T. Ohki, E. Yanase and Y. Ueno, Bioorg. Med. Chem. Lett., 2012, 22, 253–257 CrossRef CAS PubMed; (h) D. W. Dodd and R. H. E. Hudson, Mini-Rev. Org. Chem., 2009, 6, 378–379 CrossRef CAS; (i) M. L. Capobianco, A. Cazzato, S. Alesi and G. Barbarella, Bioconjugate Chem., 2007, 19, 171–177 CrossRef PubMed; (j) D. M. Kolpashchikov, Chem. Rev., 2010, 110, 4709–4723 CrossRef CAS PubMed; (k) A. Okamoto, K. Tainaka and I. Saito, J. Am. Chem. Soc., 2003, 125, 4972–4973 CrossRef CAS PubMed.
- For reviews, see: (a) M. E. Hawkins, Cell Biochem. Biophys., 2001, 34, 257–281 CrossRef CAS PubMed; (b) N. Dai and E. T. Kool, Chem. Soc. Rev., 2011, 40, 5756–5770 RSC; (c) Y. N. Teo and E. T. Kool, Chem. Rev., 2012, 112, 4221–4245 CrossRef CAS PubMed.
- Selected examples: (a) T. Ono, S. Wang, C.-K. Koo, L. Engstrom, S. S. David and E. T. Kool, Angew. Chem., Int. Ed., 2012, 51, 1689–1692 CrossRef CAS PubMed; (b) J. W. Jung, S. K. Edwards and E. T. Kool, ChemBioChem, 2013, 14, 440–444 CrossRef CAS PubMed; (c) J. Riedl, P. Ménová, R. Pohl, P. Orság, M. Fojta and M. Hocek, J. Org. Chem., 2012, 77, 8287–8293 CrossRef CAS PubMed; (d) J. Riedl, R. Pohl, N. P. Ernsting, P. Orság and M. Fojta, Chem. Sci., 2012, 3, 2797–2806 RSC; (e) O. Köhler, D. V. Jarikote, I. Singh, V. S. Parmar, E. Weinhold and O. Seitz, Pure Appl. Chem., 2005, 77, 327–338 Search PubMed.
- (a) M. J. Davies, A. Shah and I. J. Bruce, Chem. Soc. Rev., 2000, 29, 97–107 RSC; (b) L. M. Wilhelmsson, Q. Rev. Biophys., 2010, 43, 159–183 CrossRef CAS PubMed; (c) R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010 Search PubMed; (d) M. Rist and J. Marino, Curr. Org. Chem., 2002, 6, 775–793 CrossRef CAS; (e) J. N. Wilson and E. T. Kool, Org. Biomol. Chem., 2006, 4, 4265–4274 RSC; (f) A. A. Tanpure, M. G. Pawar and S. G. Srivatsan, Isr. J. Chem., 2013, 53, 366–378 CrossRef CAS; (g) For RNA series, see: S. G. Srivatsan and A. A. Sawant, Pure Appl. Chem., 2010, 83, 213–232 Search PubMed.
- (a) N. J. Greco and Y. Tor, J. Am. Chem. Soc., 2005, 127, 10784–10785 CrossRef CAS PubMed; (b) N. J. Greco and Y. Tor, Tetrahedron, 2007, 63, 3515–3527 CrossRef CAS PubMed; (c) D. Shin, R. W. Sinkeldam and Y. Tor, J. Am. Chem. Soc., 2011, 133, 14912–14915 CrossRef CAS PubMed; (d) J. Riedl, R. Pohl, L. Rulíšek and M. Hocek, J. Org. Chem., 2012, 77, 1026–1044 CrossRef CAS PubMed; (e) T. Pesnot, L. M. Tedaldi, P. G. Jambrina, E. Rosta and G. K. Wagner, Org. Biomol. Chem., 2013, 11, 6357–6371 RSC; (f) Y. Saito, A. Suzuki, Y. Okada, Y. Yamasaka, N. Nemoto and I. Saito, Chem. Commun., 2013, 49, 5684–5686 RSC; (g) Y. Saito, A. Suzuki, S. Ishioroshi and I. Saito, Tetrahedron Lett., 2011, 52, 4726–4729 CrossRef CAS; (h) A. A. Tanpure and S. G. Srivatsan, Chem.–Eur. J., 2011, 17, 12820–12827 CrossRef CAS PubMed; (i) A. Okamoto, K. Tainaka and Y. Fujiwara, J. Org. Chem., 2006, 71, 3592–3598 CrossRef CAS PubMed; (j) N. Ben Gaied, N. Glasser, N. Ramalanjaona, H. Beltz, P. Wolff, R. Marquet, A. Burger and Y. Mély, Nucleic Acids Res., 2005, 33, 1031–1039 CrossRef CAS PubMed; (k) T. Kanamori, H. Ohzeki, Y. Masaki, A. Ohkubo, M. Takahashi, K. Tsuda, T. Ito, M. Shirouzu, K. Kuwasako, Y. Muto, M. Sekine and K. Seio, ChemBioChem, 2015, 16, 167–176 CrossRef CAS PubMed.
- (a) A. P. Demchenko, J. Fluoresc., 2010, 20, 1099–1128 CrossRef PubMed; (b) A. P. Demchenko, J. Mol. Struct., 2014, 1077, 51–67 CrossRef CAS.
- For an example, see: L. Xu, M.-L. He, H.-B. Yang and X. Qian, Dalton Trans., 2013, 42, 8218–8222 RSC.
- Selected examples: (a) P. A. Hopkins, R. W. Sinkeldam and Y. Tor, Org. Lett., 2014, 16, 5290–5293 CrossRef CAS PubMed; (b) A. Suzuki, K. Kimura, S. Ishioroshi, I. Saito, N. Nemoto and Y. Saito, Tetrahedron Lett., 2013, 54, 2348–2352 CrossRef CAS.
- Selected example: (a) S. Sasaki, Y. Niko, A. S. Klymchenko and G.-I. Konishi, Tetrahedron, 2014, 70, 7551–7559 CrossRef CAS; Applications of this concept to ODNs, see: (b) A. Okamoto, K. Tainaka, K.-I. Nishiza and I. Saito, J. Am. Chem. Soc., 2005, 127, 13128–13129 CrossRef CAS PubMed; (c) A. Suzuki, N. Nemoto, I. Saito and Y. Saito, Org. Biomol. Chem., 2013, 12, 660–666 RSC; (d) A. Suzuki, T. Yanaba, I. Saito and Y. Saito, ChemBioChem, 2014, 15, 1638–1644 CrossRef CAS PubMed; (e) H.-I. Un, C.-B. Huang, C. Huang, T. Jia, X.-L. Zhao, C.-H. Wang, L. Xu and H.-B. Yang, Org. Chem. Front., 2014, 1, 1083–1090 RSC.
- (a) A. P. Demchenko, Trends Biotechnol., 2005, 23, 456–460 CrossRef CAS PubMed; (b) A. P. Demchenko, FEBS Lett., 2006, 580, 2951–2957 CrossRef CAS PubMed; (c) Z. Xu, L. Xu, J. Zhou, Y. Xu, W. Zhu and X. Qian, Chem. Commun., 2012, 48, 10871–10873 RSC.
- P. K. Sengupta and M. Kasha, Chem. Phys. Lett., 1979, 68, 382–385 CrossRef CAS.
- (a) P. T. Chou, M. L. Martinez and J. H. Clements, J. Phys. Chem., 1993, 97, 2618–2622 CrossRef CAS; (b) T. C. Swinney and D. F. Kelley, J. Chem. Phys., 1993, 99, 211–221 CrossRef CAS.
- A. S. Klymchenko and A. P. Demchenko, Phys. Chem. Chem. Phys., 2003, 5, 461–468 RSC.
- N. Chattopadhyay, M. Barroso, C. Serpa, L. G. Arnaut and S. J. Formosinho, Chem. Phys. Lett., 2004, 387, 258–262 CrossRef CAS.
- (a) M. Sarkar, J. G. Ray and P. K. Sengupta, Spectrochim. Acta, Part A, 1996, 52, 275–278 CrossRef; (b) A. S. Klymchenko and A. P. Demchenko, Langmuir, 2002, 18, 5637–5639 CrossRef CAS.
- (a) A. S. Klymchenko, G. Duportail, Y. Mély and A. P. Demchenko, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11219–11224 CrossRef CAS PubMed; (b) A. S. Klymchenko, Y. Mély, A. P. Demchenko and G. Duportail, Biochim. Biophys. Acta, Biomembr., 2004, 1665, 6–19 CrossRef CAS PubMed; (c) V. V. Shynkar, A. S. Klymchenko, C. Kunzelmann, G. Duportail, C. D. Muller, A. P. Demchenko, J.-M. Freyssinet and Y. Mély, J. Am. Chem. Soc., 2007, 129, 2187–2193 CrossRef CAS PubMed; (d) S. Oncul, A. S. Klymchenko, O. A. Kucherak, A. P. Demchenko, S. Martin, M. Dontenwill, Y. Arntz, P. Didier, G. Duportail and Y. Mély, Biochim. Biophys. Acta, Biomembr., 2010, 1798, 1436–1443 CrossRef CAS PubMed; (e) G. M'Baye, A. S. Klymchenko, D. A. Yushchenko, V. V. Shvadchak, T. Ozturk, Y. Mély and G. Duportail, Photochem. Photobiol. Sci., 2007, 6, 71–76 CrossRef PubMed.
- V. V. Shynkar, A. S. Klymchenko, G. Duportail, A. P. Demchenko and Y. Mély, Biochim. Biophys. Acta, 2005, 1712, 128–136 CrossRef CAS PubMed.
- (a) S. Ercelen, A. S. Klymchenko and A. P. Demchenko, FEBS Lett., 2003, 538, 25–28 CrossRef CAS PubMed; (b) A. S. Klymchenko, S. V. Avilov and A. P. Demchenko, Anal. Biochem., 2004, 329, 43–57 CrossRef CAS PubMed; (c) V. Y. Postupalenko, V. V. Shvadchak, G. Duportail, V. G. Pivovarenko, A. S. Klymchenko and Y. Mély, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 424–432 CrossRef CAS PubMed; (d) V. V. Shvadchak, A. S. Klymchenko, H. de Rocquigny and Y. Mély, Nucleic Acids Res., 2009, 37, e25 CrossRef PubMed; (e) A. V. Strizhak, V. Y. Postupalenko, V. V. Shvadchak, N. Morellet, E. Guit- tet, V. G. Pivovarenko, A. S. Klymchenko and Y. Mély, Bioconjugate Chem., 2012, 23, 2434–2443 CrossRef CAS PubMed.
- A. S. Klymchenko, V. V. Shvadchak, D. A. Yushchenko, N. Jain and Y. Mély, J. Phys. Chem. B, 2008, 112, 12050–12055 CrossRef CAS PubMed.
- B. Sengupta, S. M. Reilly, D. E. Davis, K. Harris, R. M. Wadkins, D. Ward, D. Gholar and C. Hampton, J. Phys. Chem. B, 2014, 119, 2546–2556 CrossRef PubMed.
- (a) D. Dziuba, V. Y. Postupalenko, M. Spadafora, A. S. Klymchenko, V. Guérineau, Y. Mély, R. Benhida and A. Burger, J. Am. Chem. Soc., 2012, 134, 10209–10213 CrossRef CAS PubMed; (b) A. A. Kuznetsova, N. A. Kuznetsov, Y. N. Vorobjev, N. P. F. Barthes, B. Y. Michel, A. Burger and O. S. Fedorova, PLoS One, 2014, 9, e100007 CrossRef PubMed.
- M. Spadafora, V. Y. Postupalenko, V. V. Shvadchak, A. S. Klymchenko, Y. Mély and A. Burger, Tetrahedron, 2009, 65, 7809–7816 CrossRef CAS.
- D. Dziuba, I. A. Karpenko, N. P. F. Barthes, B. Y. Michel, A. S. Klymchenko, R. Benhida, A. P. Demchenko, Y. Mély and A. Burger, Chem.–Eur. J., 2014, 20, 1998–2009 CrossRef PubMed.
- (a) A. P. Demchenko, K.-C. Tang and P.-T. Chou, Chem. Soc. Rev., 2013, 42, 1379–1408 RSC; (b) V. I. Tomin, A. P. Demchenko and P.-T. Chou, J. Photochem. Photobiol., C, 2015, 22, 1–18 CrossRef CAS.
- R. Chinchilla and C. Najera, Chem. Rev., 2007, 107, 874–922 CrossRef CAS PubMed.
- (a) G. T. Hwang, Y. J. Seo and B. H. Kim, Tetrahedron Lett., 2005, 46, 1475–1477 CrossRef CAS; (b) Y. Saito, K. Hanawa, K. Motegi, K. Omoto and A. Okamoto, Tetrahedron Lett., 2005, 46, 7605–7608 CrossRef CAS; (c) Y. Saito, Y. Miyauchi, A. Okamoto and I. Saito, Chem. Commun., 2004, 1704–1705 RSC; (d) A. Okamoto, K. Kanatani and I. Saito, J. Am. Chem. Soc., 2004, 126, 4820–4827 CrossRef CAS PubMed.
- J. H. Ryu, Y. J. Seo, G. T. Hwang, J. Y. Lee and B. H. Kim, Tetrahedron, 2007, 63, 3538–3547 CrossRef CAS.
- Q. Xiao, R. T. Ranasinghe, A. M. P. Tang and T. Brown, Tetrahedron, 2007, 63, 3483–3490 CrossRef CAS.
- L. A. Agrofoglio, I. Gillaizeau and Y. Saito, Chem. Rev., 2003, 103, 1875–1916 CrossRef CAS PubMed.
- S. Gobbi, A. Rampa, A. Bisi, F. Belluti, L. Piazzi, P. Valenti, A. Caputo, A. Zampiron and M. Carrara, J. Med. Chem., 2003, 46, 3662–3669 CrossRef CAS PubMed.
- D. Dziuba, R. Benhida and A. Burger, Synthesis, 2011, 2159–2164 CAS.
- (a) J.-I. Asakura and M. J. Robins, Tetrahedron Lett., 1988, 29, 2855–2858 CrossRef CAS; (b) J. Asakura and M. J. Robins, J. Org. Chem., 1990, 55, 4928–4933 CrossRef CAS.
- (a) F. W. Hobbs Jr., J. Org. Chem., 1989, 54, 3420–3422 CrossRef; (b) M. J. Robins, R. S. Vinayak and S. G. Wood, Tetrahedron Lett., 1990, 31, 3731–3734 CrossRef CAS.
- (a) M. J. Robins and P. J. Barr, Tetrahedron Lett., 1981, 22, 421–424 CrossRef CAS; (b) M. J. Robins and P. J. Barr, J. Org. Chem., 1983, 48, 1854–1862 CrossRef CAS; (c) E. De Clercq, J. Descamps, J. Balzarini, J. Giziewicz, P. J. Barr and M. J. Robins, J. Med. Chem., 1983, 26, 661–666 CrossRef CAS PubMed; (d) N. Esho, J.-P. Desaulniers, B. Davies, H. M. P. Chui, M. S. Rao, C. S. Chow, S. Szafert and R. Dembinski, Bioorg. Med. Chem., 2005, 13, 1231–1238 CrossRef CAS PubMed.
- From 5-ethynyluridines: (a) G. Luoni, C. McGuigan, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, Bioorg. Med. Chem. Lett., 2005, 15, 3791–3796 CrossRef CAS PubMed; (b) C. McGuigan, O. Bidet, M. Derudas, G. Andrei, R. Snoeck and J. Balzarini, Bioorg. Med. Chem., 2009, 17, 3025–3027 CrossRef CAS PubMed.
- From 5-iodouridines: (a) C. McGuigan, C. J. Yarnold and G. Jones, J. Med. Chem., 1999, 42, 4479–4484 CrossRef CAS PubMed; (b) S. Srinivasan, C. McGuigan, G. Andrei and R. Snoeck, Bioorg. Med. Chem. Lett., 2001, 11, 391–393 CrossRef CAS PubMed; (c) C. McGuigan, A. Brancale, G. Andrei and R. Snoeck, Bioorg. Med. Chem. Lett., 2003, 13, 4511–4513 CrossRef CAS PubMed; (d) A. Brancale, C. McGuigan, G. Andrei and R. Snoeck, Bioorg. Med. Chem. Lett., 2000, 10, 1215–1217 CrossRef CAS PubMed; (e) C. McGuigan, M. Derudas, M. Quintiliani, G. Andrei, R. Snoeck, G. Henson and J. Balzarini, Bioorg. Med. Chem. Lett., 2009, 19, 6264–6267 CrossRef CAS PubMed; (f) A. Angell, C. McGuigan, L. Garcia Sevillano, R. Snoeck, G. Andrei, E. De Clercq and J. Balzarini, Bioorg. Med. Chem. Lett., 2004, 14, 2397–2399 CrossRef CAS PubMed; (g) A. Brancale, C. McGuigan, B. Algain, P. Savy, R. Benhida, J. L. Fourrey, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, Bioorg. Med. Chem. Lett., 2001, 11, 2507–2510 CrossRef CAS PubMed.
- For a review, see: (a) A. Brancale, S. Srinivasan, C. McGuigan, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, Antiviral Chem. Chemother., 2000, 11, 383–393 CrossRef CAS PubMed; (b) C. McGuigan, A. Brancale, H. Barucki, S. Srinivasan, G. Jones, R. Pathirana, S. Blewett, R. Alvarez, C. J. Yarnold, A. Carangio, A. Velázquez, S. Andrei, G. Snoeck, R. De Clercq and E. Balzarini, Drugs Future, 2000, 25, 1151–1161 CrossRef CAS; (c) C. McGuigan and J. Balzarini, Antiviral Res., 2006, 71, 149–153 CrossRef CAS PubMed.
- (a) J. Haralambidis, M. Chai and G. W. Tregear, Nucleic Acids Res., 1987, 15, 4857–4876 CrossRef CAS PubMed; (b) K. A. Cruickshank and D. L. Stockwell, Tetrahedron Lett., 1988, 29, 522l–5224 Search PubMed; (c) G. T. Crisp and B. L. Flynn, J. Org. Chem., 1993, 58, 6614–6619 CrossRef CAS; (d) M. J. Robins, K. Miranda, V. K. Rajwanshi, M. A. Peterson, G. Andrei, R. Snoeck, E. De Clercq and J. Balzarini, J. Med. Chem., 2006, 49, 391–398 CrossRef CAS PubMed; (e) E. Petricci, M. Radi, F. Corelli and M. Botta, Tetrahedron Lett., 2003, 44, 9181–9184 CrossRef CAS.
- (a) N. E. Leadbeater and B. J. Tominack, Tetrahedron Lett., 2003, 44, 8653–8656 CrossRef CAS; (b) J.-H. Kim, D.-H. Lee, B.-H. Jun and Y.-S. Lee, Tetrahedron Lett., 2007, 48, 7079–7084 CrossRef CAS; (c) A. Tougerti, S. Negri and A. Jutand, Chem.–Eur. J., 2007, 13, 666–676 CrossRef CAS PubMed; (d) D. Gelman and S. L. Buchwald, Angew. Chem., Int. Ed., 2003, 42, 5993–5996 CrossRef CAS PubMed; (e) O. R'kyek, N. Halland, A. Lindenschmidt, J. Alonso, P. Lindemann, M. Urmann and M. Nazaré, Chem.–Eur. J., 2010, 16, 9986–9989 CrossRef PubMed; (f) P. Arsenyan, K. Rubina, J. Vasiljeva and S. Belyakov, Tetrahedron Lett., 2013, 54, 6524–6528 CrossRef CAS; (g) P. Y. Choy, W. K. Chow, C. M. So, C. P. Lau and F. Y. Kwong, Chem.–Eur. J., 2010, 16, 9982–9985 CrossRef CAS PubMed.
- E. S. Kumarasinghe, M. A. Peterson and M. J. Robins, Tetrahedron Lett., 2000, 41, 8741–8745 CrossRef CAS.
- A. S. Klymchenko, V. G. Pivovarenko and A. P. Demchenko, Spectrochim. Acta, Part A, 2003, 59, 787–792 CrossRef.
- C. Reichardt, Chem. Rev., 1994, 94, 2319–2358 CrossRef CAS.
- A. S. Klymchenko and A. P. Demchenko, New J. Chem., 2004, 28, 687–692 RSC.
- A. S. Klymchenko, T. Ozturk, V. G. Pivovarenko and A. P. Demchenko, Can. J. Chem., 2001, 79, 358–363 CrossRef CAS.
- C. Boudier, A. S. Klymchenko, Y. Mély and A. Follenius-Wund, Photochem. Photobiol. Sci., 2009, 8, 814–821 CrossRef CAS PubMed.
- (a) S. M. Ormson, R. G. Brown, F. Vollmer and W. Rettig, J. Photochem. Photobiol., A, 1994, 81, 65–72 CrossRef CAS; (b) O. A. Kucherak, L. Richert, Y. Mély and A. S. Klymchenko, Phys. Chem. Chem. Phys., 2012, 14, 2292–2300 RSC.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- (a) E. Z. Lippert, Z. Naturforsch., 1955, 10, 541–545 CrossRef; (b) N. Mataga, Y. Kaifu and M. Koizumi, Bull. Chem. Soc. Jpn., 1956, 29, 465–470 CrossRef CAS.
- C. A. Kenfack, A. S. Klymchenko, G. Duportail, A. Burger and Y. Mély, Phys. Chem. Chem. Phys., 2012, 14, 8910–8918 RSC.
- C.-C. Hsieh, C.-M. Jiang and P.-T. Chou, Acc. Chem. Res., 2010, 43, 1364–1374 CrossRef CAS PubMed.
- O. M. Zamotaiev, V. Y. Postupalenko, V. V. Shvadchak, V. G. Pivovarenko, A. S. Klymchenko and Y. Mély, Bioconjugate Chem., 2010, 22, 101–107 CrossRef PubMed.
- (a) A. S. Klymchenko, V. G. Pivovarenko, T. Ozturk and A. P. Demchenko, New J. Chem., 2003, 27, 1336–1343 RSC; (b) V. V. Shynkar, Y. Mely, G. Duportail, E. Piémont, A. S. Klymchenko and A. P. Demchenko, J. Phys. Chem. A, 2003, 107, 9522–9529 CrossRef CAS.
- A. J. G. Strandjord and P. F. Barbara, J. Phys. Chem., 1985, 89, 2355–2361 CrossRef CAS.
- (a) K. Kipper, K. Herodes and I. Leito, J. Chromatogr. A, 2011, 1218, 8175–8180 CrossRef CAS PubMed; (b) K. Kipper, K. Herodes, I. Leito and L. Nei, Analyst, 2011, 136, 4587–4594 RSC.
- R. A. Moore, J. Lee and G. W. Robinson, J. Phys. Chem., 1985, 89, 3648–3654 CrossRef CAS.
- (a) For instance, see 2,6-diaminopurine: D. C. Ward, E. Reich and L. Stryer, J. Biol. Chem., 1969, 244, 1228–1237 CrossRef CAS PubMed; (b) For a recent example of a bright solvatochromic dye in water, see: Y. Niko, Y. Cho, S. Kawauchi and G.-I. Konishi, RSC Adv., 2014, 4, 36480–36484 RSC.
- (a) V. G. Pivovarenko, O. M. Zamotaiev, V. V. Shvadchak, V. Y. Postupalenko, A. S. Klymchenko and Y. Mély, J. Phys. Chem. A, 2012, 116, 3103–3109 CrossRef CAS PubMed; (b) R. Das, A. S. Klymchenko, G. Duportail and Y. Mély, Photochem. Photobiol. Sci., 2009, 8, 1583–1589 CrossRef CAS PubMed.
- (a) V. V. Shynkar, A. S. Klymchenko, E. Piémont, A. P. Demchenko and Y. Mély, J. Phys. Chem. A, 2004, 108, 8151–8159 CrossRef CAS; (b) W. Caarls, M. S. Celej, A. P. Demchenko and T. M. Jovin, J. Fluoresc., 2010, 20, 181–190 CrossRef CAS PubMed.
- K. Rurack and U. Resch-Genger, Chem. Soc. Rev., 2002, 31, 116–127 RSC.
- A. P. Demchenko, Anal. Biochem., 2005, 343, 1–22 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The experimental protocols for the synthesis and characterization, the 1H and 13C NMR spectra of the compounds 2–4 and 9–24, and the HPLC of the final products 2–4 can be consulted in the ESI. See DOI: 10.1039/c5ra02709h |
| ‡ Present address: Ecole Polytechnique Fédérale de Lausanne, Institute of Chemical Sciences and Engineering, Laboratory of Protein Engineering, BCH, Av. Forel 2, CH-1015 Lausanne, Switzerland. |
| § Present address: Institute of Organic Chemistry and Biochemistry, Academy of Sciences of the Czech Republic, Flemingovo nam. 2, CZ-16610, Prague 6, Czech Republic. |
| ¶ An one-pot two-step procedure, consisting in Pd-catalyzed coupling followed by the removal of the silyl group, was also attempted; however, a moderate yield of 64% was obtained. |
| || Concerning the furano[2,3-d]pyrimidin-2-one nucleoside 20, NMR and MS analysis gave the spectroscopic characteristics consistent with a 5-endo cyclized derivative. Indeed, the disappearance in 1H-NMR of the imide NH signal at 9.35 ppm was observed in favor of a new singlet at 6.92 ppm corresponding to the proton of the fused furan ring. Moreover in 13C-NMR, the lost of the two typical signals of the ethynyl carbons at 83 and 87 ppm in favor of two new aromatic peaks respectively at 99 and 146 ppm bring the undeniable evidence of a furanopyrimidine scaffold. It is noteworthy that the deshielding (>10 ppm) of C-4 and C-5 of uracil fragment to respectively 107 ppm and 171 constitutes another proof of the proposed bicyclic aromatic structure. |
| This journal is © The Royal Society of Chemistry 2015 |