
Effects of magnetic ordering and electron correlations on the stability of FeN
Zhonglong Zhao,
Kuo Bao,
Defang Duan,
Fubo Tian,
Bingbing Liu and
Tian Cui*
State Key Laboratory of Superhard Materials, College of Physics, Jilin University, Changchun 130012, People's Republic of China. E-mail: cuitian@jlu.edu.cn; Fax: +86-431-85168825; Tel: +86-431-85168825
First published on 19th March 2015
Abstract
Iron mononitride has attracted much interest because of its interesting magnetoelectric properties. However, whether the ground state of FeN has a rock-salt (rs) or a zinc-blende (zb) structure is still controversial. Clarification of this issue has been impeded by the complex magnetic ordering and strong electron correlation effects. Here, we study the relative stability of rs and zb FeN toward different spin orderings (ferromagnetic, antiferromagnetic, and paramagnetic) at pressures of 0–100 GPa, with the GGA-PBE, LDA+U, and HSE hybrid exchange–correlation functionals. We find that the competition between direct and indirect exchange interactions can drive magnetic structure phase transitions for rs-FeN at high pressures, whereas zb-FeN is still nonmagnetic. Strikingly, the energy difference between rs and zb FeN decreases and finally vanishes as the occupied minority-spin t2g orbitals of rs-FeN are depleted when 3d electron correlations are considered. These results demonstrate that an appropriate treatment of electron correlations is important for determining the stability and properties of 3d transition metal nitrides.
1. Introduction
Iron mononitride (FeN) is of great interest because of its promising applications in magnetic, spintronic, and mechanical devices.1 However, the ground state structure for FeN is still an open question.2–5 Two kinds of FeN have been reported experimentally. One is the rocksalt (rs) structure with antiferromagnetic (AF) ordering.2,3 The other is the zinc-blende (zb) structure that shows micromagnetic properties.4 According to recent studies, film samples can be prepared for both phases with physical vapor deposition methods.6–10 Theoretical studies have attempted to clarify the magnetism and relative stability of rs and zb FeN, but with controversial results. For example, Shimizu et al.11 and Houari et al.1 showed that ferromagnetic (FM) rs-FeN is more stable than non-magnetic (NM) zb-FeN. In contrast, Kong proposed AF1 rs-FeN with magnetic ordering consisting of alternating single ferromagnetic sheets along the [001] direction.12 Lukashev et al.13 and Soni et al.14 predicted that NM zb-FeN is the ground state and will transform into rs-FeN at high pressures. Most recent first-principles studies have also supported the NM zb ground state for FeN with high hardness.15,16 In addition to the ground state controversy, there is a large difference between the estimated lattice parameters and experimental reports for rs-FeN, although the experimental lattice of zb-FeN can be reproduced.1,3,4From a theoretical point of view, clarification of the ground state of FeN is mainly prevented by the complex magnetic ordering. Although configurations such as AF1 and AF2 (alternating single ferromagnetic sheets along the [111] direction) have been investigated for the rs phase, there is no study that considers a complete set of magnetic orderings. On the other hand, the accuracy of density functional theory (DFT) for period 4 transition metal (TM) compounds is decreased by the strong correlations between 3d electrons, and the normal exchange–correlation functional predicts incorrect metallic ground states for many insulating TM oxides, such as MnO, FeO, CoO, and NiO.17–19 Furthermore, a unique structural anomaly of rs and zb MnN caused by strong 3d electron correlations has been found.20 To our knowledge, the effects of electron correlations on the stability of FeN remain unclear.
In this work, we study the effect of different magnetic orderings and electron correlations on the stability of FeN at 0–100 GPa by using ab initio calculations. The NM, FM, five AF orderings (AF1 to AF5), and a paramagnetic (PM) state were constructed. To consider the electron correlations, the stability of FeN toward different exchange–correlation functionals, such as GGA-PBE, LDA+U, and the HSE hybrid functional, was compared. The competition between the direct magnetic exchange and the superexchange interactions can drive the magnetic phase transition for rs-FeN at high pressure. Moreover, the relative energy of rs and zb FeN changed for different exchange–correlation functionals. Our results help clarify the ground state controversy of FeN and highlight the importance of different magnetic orderings and electron correlations in determining the structure and properties of 3d TM mononitrides.
2. Computational details
First-principles calculations are performed by using the Vienna ab initio simulation package VASP code21 with the projector augmented waves (PAW) method.22 For the exchange–correlation energy, the Perdew–Burke–Ernzerhof version of the generalized gradient approximation (GGA-PBE),23 a combination of LDA24 with a Hubbard Coulomb term (LDA+U) method,25,26 and the Heyd–Scuseria–Ernzerhof (HSE) hybrid functional27–29 are used. We employ the rotationally invariant approach introduced by Dudarev et al.26 for the LDA+U calculations with
 | (1) |
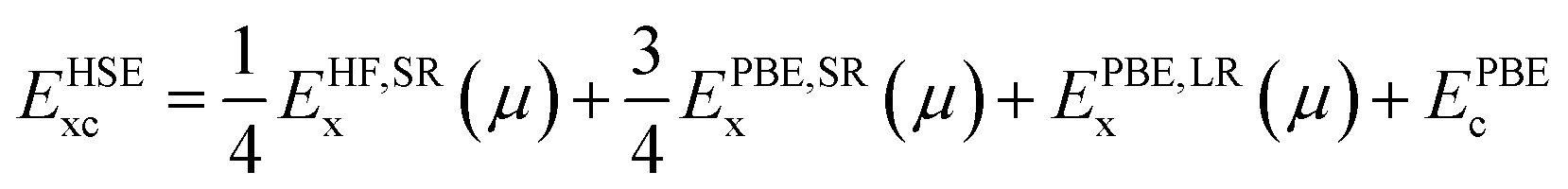 | (2) |
In this form, the normal exchange term of the GGA-PBE functional is split into two parts: a short-range (SR) and a long-range (LR) term. In the calculations, quarter of the short-range part is replaced with a short-range Hartree–Fock term with a screening length of 0.2 Å−1. The correlation part of the GGA-PBE is unchanged. By performing accurate convergence tests, the plane-wave basis set cutoff is chosen as 520 eV, and a uniform k-grid mesh of 0.03 × 2π Å−1 within the Monkhorst-Pack scheme is set for sampling the Brillouin zone during the PBE, LDA+U, and HSE calculations. This ensures that the total energy is well converged to better than 1 meV per atom. These settings are tested to be adequate to ensure that the magnetic moments are well converged to less than 0.01 μB/Fe.
3. Results and discussion
The magnetic configurations can be classified by the orientation of Fe atomic moments on the face-centered cubic sublattice of both the rs and zb FeN structures. The five AF orderings (AF1 to AF5, inset of Fig. 1) in addition to the FM ordering are studied for comparing the energies. The AF orderings considered are the same as those investigated for MnS and MnO,30,31 which can be distinguished by the stacking in layers of opposite spins. In other words, AF1 and AF2 are characterized by a monolayer alternation of spins in the [100] and [111] directions of the conventional 8-atom cubic cell, whereas AF3, AF4, and AF5 represent a bilayer alternation of spins in the [210], [110], and [100] directions, respectively. Generally, considering the Kramers–Anderson superexchange32,33 in addition to the direct magnetic exchange is important for constructing a complete set of AF couplings. In this respect, the above AF spin arrangements can be classified according to the numbers of spin-parallel (s↑↑) and spin-antiparallel (s↑↓) interactions with their nearest neighbors (nn) and next-nearest neighbors (nnn) (Table 1), as first described by Anderson in 1950.34 Taking the ideal ionic valency for rs and zb FeN, three kinds of Fe3+(3d5)–N3−(2p6)–Fe3+(3d5) linkages may involve the superexchange and can be distinguished with different bond angles (180°, 109.47°, and 90°). Accordingly, the AF1 and AF3 orderings are the cases favoring the 109.47° superexchange of zb-FeN because it has the largest number of nn–s↑↓ interactions, whereas for the AF2 and AF4 orderings that have a larger number of nnn–s↑↓ interactions, the 180° superexchange of rs-FeN can be considered. Because the 90° Fe–N–Fe superexchange favors the FM interaction between cations,35 the AF5 ordering with 67% nn–s↑↑ interactions can be tested (Table 1 and Fig. 1). | ||
| Fig. 1 Enthalpy difference–pressure (Hdiff–P) diagram of rs-FeN with different magnetic orderings of zb-FeN at 0–100 GPa calculated with the PBE functional. Top inset shows the antiferromagnetic orderings considered (AF1 to AF5). The large spheres with different colors and arrows represent Fe atoms with positive and negative spins. N atoms are shown as small spheres. | ||
| Order | AF1 | AF2 | AF3 | AF4 | AF5 |
|---|---|---|---|---|---|
| nn–s↑↑ | 4 | 6 | 4 | 6 | 8 |
| nn–s↑↓ | 8 | 6 | 8 | 6 | 4 |
| nnn–s↑↑ | 6 | 0 | 4 | 2 | 4 |
| nnn–s↑↓ | 0 | 6 | 2 | 4 | 2 |
The enthalpy difference-pressure phase diagram of FeN calculated with the PBE functional is shown in Fig. 1. zb-FeN is the ground state, which is consistent with previous studies.4,13,15,16,36 All the assumed initial magnetic moments for zb-FeN collapsed to zero after full electronic and structural optimizations. Thus, we can conclude that it is nonmagnetic. For rs-FeN, competition between different magnetic orderings can be identified. According to Fig. 1, the energy sequence of AF1 < AF3 < AF4 < FM < AF5 < AF2 < NM at 0 GPa and the crossover between AF1 and AF4 at pressures above ∼5 GPa can be detected. PBE describes the magnetic orderings of rs-FeN as high-spin states with the calculated magnetic moment sequence in AF2 (1.75 μB/Fe) < AF4 (1.92 μB/Fe) < AF5 (1.99 μB/Fe) < AF3 (2.38 μB/Fe) < FM (2.45 μB/Fe) < AF1 (2.93 μB/Fe) at 0 GPa. In contrast to typical magnetic materials, such as rs-type MnS and MnO, with lowest-energy AF2 ordering arising from the largest number of nnn–s↑↓ due to the strong 180° superexchange interactions,30,31 AF1 ordering with a largest number of nn–s↑↓ and the minimum number of nnn–s↑↓ is favored for rs-FeN at low pressures (Fig. 1). This result implies that the origin of the AF coupling in rs-FeN at low pressures is the direct exchange interactions; the superexchange is weak for the bridging N3− anions. On these grounds, the AF3 ordering with the same direct exchange interactions, which has the same largest number of nn–s↑↓, and the additional 33% indirect exchange interactions are reasonably the second lowest in energy.
The AF1 and AF3 orderings favored at low pressure become unfavored states at high pressures. The AF4 ordering becomes the most stable ordering at ∼5 GPa for rs-FeN and the overall stable state for FeN at ∼46 GPa. This is a result of the competition between the direct and indirect exchange interactions at a shorter Fe–N distance. The stability of the AF4 ordering with a 50% s↑↑, 50% s↑↓ nn and 67% nnn–s↑↓ suggests that the indirect superexchange is important at high-pressures, which may be attributed to a bigger overlap of the wave functions between the positive and negative ions. In fact, the energy of the AF2 ordering decreased toward the AF3, AF1, and finally the FM orderings at increased pressures (Fig. 1), which may indicate that the strong 180° superexchange interactions are favored for rs-FeN at very high pressures, although this is beyond the scope of this work. We note that high pressure can significantly reduce the magnetic moments of rs-FeN with little effect on the magnetic moment sequence. For example, the magnetic moments of the AF1 (1.42 μB/Fe) and AF3 (1.38 μB/Fe) orderings favored by direct exchange are still larger than that of the AF4 (0.73 μB/Fe) ordering favored by indirect exchange at 50 GPa.
We have proved that the local spin orderings are the main factors affecting the stability of rs-FeN. However, the magnetic disturbance is weak compared with the relative energy, and the ground state of FeN is the zb phase (Fig. 1). Therefore, inadequate consideration of the magnetic ordering cannot be solely responsible for the discrepancy in the FeN ground state. The normal functionals, such as GGA-PBE and LDA, give inadequate descriptions of the exchange–correlation effects of strongly localized 3d electrons. Consequently, the band gap of some TM oxides, such as MnO, FeO, CoO, and NiO, is substantially underestimated in normal DFT calculations.17,18 Furthermore, recent studies suggest that the GGA-PBE/LDA functionals cannot even predict the correct ground state for correlated electronic materials such as MnN and MnO.20,31
To cover possible electron correlation effects, the relative stability of rs (FM and AF1 orderings) and zb FeN toward different functionals at 0–100 GPa are shown in Fig. 2. By increasing U in LDA+U calculations, the relative enthalpies of rs-FeN dropped dramatically. Significantly, the rs structure ground state can be predicted for FeN when the U values are between 3 and 4 eV. High pressure weakens the correlation effect significantly and only minor energy changes can be detected at 100 GPa for different U values. The transition of the ground state from zb-FeN to rs-FeN within LDA+U is further confirmed by HSE calculations. According to Fig. 2, the zb ground state predicted by PBE and LDA+U (U ≤ 3 eV) is ∼0.54 eV per f.u. higher in energy than rs-FeN at 0 GPa for the HSE calculations.
 | ||
| Fig. 2 Enthalpy difference–pressure (Hdiff–P) diagram of rs-FeN for FM and AF1 magnetic orderings of zb-FeN at 0–100 GPa calculated with PBE, LDA+U (U = 2, 3, 4, 5, and 6 eV), and HSE functionals. | ||
The total and orbital projected partial electronic density of states (DOS) of FeN for different exchange–correlation functionals are shown in Fig. 3. Both rs and zb phases are metallic with a primary contribution of Fe 3d electrons to the Fermi level. For zb-FeN, the electron correlation effect is weak and all three functionals give consistent DOS, even though a ∼0.03 μB/Fe moment was predicted by HSE. For rs-FeN (using FM ordering as an example), although a high-spin state (atomic magnetic moments from 2.5 to 4.2 μB/Fe for different functionals, inset of Fig. 4) makes the majority of spin t2g (↑) orbitals nearly completely filled, the filling of the minority spin t2g (↓) orbitals are different for different functionals. According to Fig. 3, the mainly occupied t2g (↓) orbitals shown by the PBE functional can be depleted to a less occupied (LDA+U, U = 4 eV) or nearly unoccupied orbitals (HSE), leaving the Fermi level in a valley between the majority and minority spin channels and transforming rs-FeN to low energies. Therefore, the underestimation of PBE of the exchange interactions among spin-up and spin-down populations may be responsible for the competition between the rs and zb ground state of FeN. Note that an analogous depletion of the intra-atomic occupied t2g (↓) orbitals is observed for the AF1 rs-FeN in the LDA+U and HSE calculations.
 | ||
| Fig. 3 Total and partial electronic DOS of FeN for PBE, LDA+U (U = 4 eV), and HSE functionals at 0 GPa. | ||
 | ||
| Fig. 4 Equilibrium lattice parameters and magnetic moments (inset) of FeN for PBE, LDA+U (U = 2, 3, 4, 5, and 6 eV), and HSE functionals. Experimental lattices are shown for comparison (ref. 2–4). | ||
The competition of the rs and zb phases revealed with different exchange–correlation functionals helps to clarify the ground state controversy of FeN in previous theoretical and experimental studies. Theoretically, the discrepancy apparently comes from using different functionals and the approach to handling the exchange–correlation effects. In this light, the atomic sphere approximation (ASA) used by Kong12 and Houari et al.1 seems to give qualitatively consistent results with the LDA+U (when U ≥ 4 eV) and HSE calculations. From an experimental point of view, the coexistence of rs and zb FeN can be understand from the tiny difference in their energies when appropriate electron correlations are taken into consideration; for example, an equivalent energy for two phases may be found between U = 3–4 eV within the LDA+U calculations (Fig. 2). In this context, FeN should be different from other strongly correlated materials, such as Mn, with an undisputed rs ground state, although the normal GGA-PBE/LDA methods have also failed to predict these.20
We finally discuss the effects of magnetic ordering and electron correlation on the lattice parameters, because most previous theoretical studies predicted a shrunken lattice for FeN. Above the Néel temperature, the magnetic stress of the disordered PM moments may be important for expanding the lattice.37 Therefore, we modeled the disorder of magnetic moments in PM rs-FeN by means of the special quasi-random structure method,37,38 using a Fe0.5↑Fe0.5↓N supercell with 48 atoms. This method has been successfully used to investigate the effect of magnetic disorder on the structure and elastic properties of CrN.37,39 The carefully selected PM configuration for FeN has a zero-spin correlation function for the first five coordination shells. According to Fig. 4, although significant changes in the lattice parameters of FeN occur for different magnetic orderings and functionals, the theoretical lattice parameter in rs-FeN is different from the experimental data by more than 8%. Our results together with previous theoretical studies,1,3,4 from the other side, prove that the magnetic stress and possible electron correlations cannot help resolve the lattice conflict, and possible surface effects in the film sample must be taken into account.
4. Conclusions
In summary, the relative stability of rs and zb FeN has been studied by a first-principles pseudopotential plane wave method considering the magnetic structures and strong electron correlation effects. The competition of the direct magnetic exchange and superexchange interactions can drive an AF1 to AF4 magnetic phase transition for rs-FeN at high pressure, whereas zb-FeN remains non-magnetic. The relative energy of rs and zb FeN changed for different exchange–correlation functionals; rs-FeN that was unfavored for PBE can be stable when using the HSE and LDA+U methods. Therefore, the ground state discrepancy of FeN with previous studies arises mainly from the treatment of correlations between Fe 3d electrons. Our results highlight the necessity of considering magnetic orderings and electron correlations in correctly describing the structural stability and properties of 3d TM compounds.Acknowledgements
This work was supported by the National Basic Research Program of China (no. 2011CB808200), Program for Changjiang Scholars and Innovative Research Team in University (no. IRT1132), National Natural Science Foundation of China (nos 51032001, 11074090, 10979001, 51025206), and National Fund for Fostering Talents of Basic Science (no. J1103202). Parts of calculations were performed in the High Performance Computing Center (HPCC) of Jilin University.References
- A. Houari, S. F. Matar, M. A. Belkhir and M. Nakhl, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 064420 CrossRef.
- H. Nakagawa, S. Nasu, H. Fujii, M. Takahashi and F. Kanamaru, Hyperfine Interact., 1992, 69, 455–458 CrossRef.
- T. Hinomura and S. Nasu, Hyperfine Interact., 1998, 111, 221–226 CrossRef CAS.
- K. Suzuki, H. Morita, T. Kaneko, H. Yoshida and H. Fujimori, J. Alloys Compd., 1993, 201, 11–16 CrossRef CAS.
- L. Rissanen, M. Neubauer, K. P. Lieb and P. Schaaf, J. Alloys Compd., 1998, 274, 74–82 CrossRef CAS.
- R. Usui, Y. Yamada and Y. Kobayashi, Hyperfine Interact., 2012, 205, 13–16 CrossRef CAS PubMed.
- R. Gupta, A. Tayal, S. M. Amir, M. Gupta, A. Gupta, M. Horisberger and J. Stahn, J. Appl. Phys., 2012, 111, 103520 CrossRef PubMed.
- A. Tayal, M. Gupta, D. M. Phase, V. R. Reddy and A. Gupta, AIP Conf. Proc., 2014, 1591, 970–971 CrossRef CAS PubMed.
- A. Tayal, M. Gupta, N. P. Lalla, A. Gupta, M. Horisberger, J. Stahn, K. Schlage and H. C. Wille, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 90, 144412 CrossRef.
- E. Z. Frątczak, J. E. Prieto and M. E. Moneta, J. Alloys Compd., 2014, 586, 375–379 CrossRef PubMed.
- H. Shimizu, M. Shirai and N. Suzuki, J. Phys. Soc. Jpn., 1998, 67, 922–926 CrossRef CAS.
- Y. Kong, J. Phys.: Condens. Matter, 2000, 12, 4161–4173 CrossRef CAS.
- P. Lukashev and W. R. L. Lambrecht, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 245205 CrossRef.
- H. R. Soni, V. Mankad, S. K. Gupta and P. K. Jha, J. Alloys Compd., 2012, 522, 106–113 CrossRef CAS PubMed.
- V. F. Hlynsson, E. Skúlason and A. L. Garden, J. Alloys Compd., 2014, 603, 172–179 CrossRef CAS PubMed.
- J. S. Chen, C. Yu and H. Lu, J. Alloys Compd., 2015, 625, 224–230 CrossRef CAS PubMed.
- K. Terakura, A. R. Williams, T. Oguchi and J. Kübler, Phys. Rev. Lett., 1984, 52, 1830–1833 CrossRef CAS.
- K. Terakura, T. Oguchi, A. R. Williams and J. Kübler, Phys. Rev. B: Condens. Matter Mater. Phys., 1984, 30, 4734–4747 CrossRef CAS.
- R. Q. Wu, G. W. Peng, L. Liu and Y. P. Feng, Appl. Phys. Lett., 2006, 89, 082504 CrossRef PubMed.
- J. A. Chan, J. Z. Liu, H. Raebiger, S. Lany and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 184109 CrossRef.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- D. M. Ceperley and B. J. Alder, Phys. Rev. Lett., 1980, 45, 566–569 CrossRef CAS.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS PubMed.
- J. Heyd and G. E. Scuseria, J. Chem. Phys., 2004, 121, 1187–1192 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef PubMed.
- R. I. Hines, N. L. Allan, G. S. Bell and W. C. Mackrodt, J. Phys.: Condens. Matter, 1997, 9, 7105 CrossRef CAS.
- A. Schron, C. Rodl and F. Bechstedt, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 165109 CrossRef.
- H. A. Kramers, Physica, 1934, 1, 182–192 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1959, 115, 2–13 CrossRef CAS.
- P. W. Anderson, Phys. Rev., 1950, 79, 350–356 CrossRef.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87–98 CrossRef CAS.
- B. Eck, R. Dronskowski, M. Takahashi and S. Kikkawa, J. Mater. Chem., 1999, 9, 1527–1537 RSC.
- B. Alling, T. Marten and I. A. Abrikosov, Nat. Mater., 2010, 9, 283–284 CrossRef CAS PubMed.
- A. Zunger, S. H. Wei, L. G. Ferreira and J. E. Bernard, Phys. Rev. Lett., 1990, 65, 353–356 CrossRef CAS.
- B. Alling, T. Marten and I. A. Abrikosov, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 184430 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |