
Microwave-assisted optimization of the manganese redox states for enhanced capacity and capacity retention of LiAlxMn2−xO4 (x = 0 and 0.3) spinel materials†
Abstract
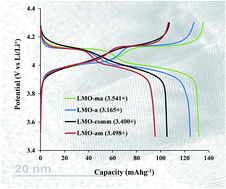
Microwave irradiation at the pre- and post-annealing steps of the synthesis of LiAlxMn2−xO4 (x = 0 and 0.3) spinel cathode materials for rechargeable lithium ion batteries is a useful strategy to optimize the average manganese valence number (nMn) for enhanced capacity and capacity retention. The strategy impacts on the lattice parameter, average manganese valence, particle size and morphology, reversibility of the de-intercalation/intercalation processes, and capacity retention upon continuous cycling. Microwave irradiation is able to shrink the particles for improved crystallinity. The XPS data clearly suggest that microwave irradiation can be used to tune the manganese valence (nMn), and that the LiAlxMn2−xO4 with nMn ≈ 3.5+ gives the best electrochemical performance. These new findings promise to revolutionize how we use microwave irradiation in the preparation of energy materials and various other materials for energy storage and conversion materials for enhanced performance.
 Please wait while we load your content...
Please wait while we load your content...

